

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://www.thejh.org |
Case Report
Volume 11, Number 4, August 2022, pages 142-147

Immune Checkpoint Inhibitor-Induced Hemophagocytic Lymphohistiocytosis in a Patient With Squamous Cell Carcinoma
Rosalyn Marara, c, Sruti Prathivadhi-Bhayankarama, Mridula Krishnanb
aDepartment of Internal Medicine, University of Nebraska Medical Center, Omaha, NE 68198, USA
bDivision of Oncology and Hematology, University of Nebraska Medical Center, Omaha, NE 68198, USA
cCorresponding Author: Rosalyn Isam Marar, Department of Internal Medicine, University of Nebraska Medical Center, Omaha, NE 68198, USA
Manuscript submitted July 15, 2022, accepted August 22, 2022, published online August 30, 2022
Short title: ICI-Induced HLH With SCC
doi: https://doi.org/10.14740/jh1033
| Abstract | ▴Top |
Programmed cell death protein 1 (PD-1) checkpoint inhibitors such as pembrolizumab are novel therapeutics used to treat various advanced malignancies and have been shown to increase patient survival in several studies. However, these drugs have a toxicity profile that ranges from mild side effects such as dermatitis to life-threatening complications. We present a case of pembrolizumab-induced hemophagocytic lymphohistiocytosis (HLH) in an 80-year-old patient with squamous cell carcinoma (SCC) of presumed cutaneous primary. This patient initially presented with weakness and pancytopenia, thought to be immune-related. She developed progressive anemia, after which further workup revealed concern for HLH. She recovered after a course of steroids, tocilizumab, and etoposide. To our knowledge, this patient’s course is among a few rare cases of immune checkpoint inhibitor (ICI)-mediated HLH. This case highlights the need for early diagnosis and recognition of HLH as a potential toxicity related to ICI therapy.
Keywords: Pembrolizumab; Hemophagocytic lymphohistiocytosis; Tocilizumab
| Introduction | ▴Top |
Pembrolizumab is an immune checkpoint protein inhibitor that targets programmed cell death protein 1 (PD-1) or programmed cell death ligand (PD-L1) [1, 2]. Many cancers display immune tolerance by downregulating the T-cell-mediated response, including the PD-1 and PD-L1 pathways [2]. Immune checkpoint inhibitors (ICIs) like pembrolizumab are designed to block these inhibitory signals and restore the natural antitumor response mediated by these regulatory T cells [3]. However, pembrolizumab may also over-activate T cells, triggering a hyper-inflammatory state such as hemophagocytic lymphohistiocytosis (HLH) [4]. Although the exact physiologic mechanism behind ICI-induced HLH is poorly understood, translational research suggests that these side effects may develop through a combination of widespread T-cell activation, production of inflammatory cytokines, defective granule-mediated cytotoxicity of T lymphocytes, and natural killer cell dysfunction [5]. The abundant release of cytokines activate macrophages to engulf red blood cells and create hemophagocytosis, the characteristic feature of HLH [6].
HLH is a life-threatening condition with a mortality rate of over 66% [7]. It is a diagnosis that requires prompt treatment to avoid progressive organ failure. Unfortunately, less than 50% of adults with HLH receive directed therapy due to a lack of awareness and missed diagnosis [8]. Patients with this disease present with recurrent high fever and laboratory tests showing pancytopenia, hyperferritinemia, hypertriglyceridemia, and hypofibrinogenemia [8]. As many of these symptoms overlap with the clinical picture of infection and sepsis, and there exists a paucity of diagnostic tools, HLH is often overlooked [9]. Recently, there have been case reports and database reviews of ICI-induced secondary HLH. However, these reports illustrate a wide variety of different diagnostic and therapeutic approaches with variable outcomes [6]. Moreover, retrospective observational cross-sectional studies have shown that the incidence of ICI-related HLH was estimated to be < 0.1% [10]. Our patient responded well after a trial of dexamethasone, tocilizumab, and etoposide. Our case highlights the importance of recognizing rare but life-threatening toxicities associated with ICI use.
| Case Report | ▴Top |
Investigations
Our case was an 80-year-old female with a past medical history significant for multiple skin cancers that had been resected over the years including a well-differentiated squamous cell carcinoma (SCC) on right and left forearm resected in 2014 and 2019, respectively. She presented to her primary care physician with swelling in her left groin with associated pain noted in February 2020. Physical examination at that time did not demonstrate swelling of any other lymph nodes and no remarkable skin findings. A computer tomography (CT) scan was concerning for pathologic appearing adenopathy in the inguinal and left pelvic lymph nodes. Lymph node biopsy demonstrated poorly differentiated non-small cell carcinoma with focal squamous differentiation (p16 negative) of unknown primary origin on immunohistochemistry. Given her pathologic pelvic lymph nodes, she underwent a pap smear and urine cytology was obtained, both negative for malignancy. It was ultimately determined she had squamous cell cancer of likely cutaneous primary given her history of multiple skin cancers.
She received treatment with local irradiation where she received 2,000 cGy in five fractions. Positive emission tomography (PET)/CT scan following radiation showed a decrease in size and metabolic activity of pelvic and inguinal lymph nodes but worsening of metastatic disease to thoracic and abdominopelvic lymph nodes, lungs, and an enlarging hypermetabolic lytic lesion involving the superior pubic ramus. She was subsequently started on pembrolizumab at 400 mg, initially every 3 weeks and later transitioned to every 6 weeks. She was noted to have progressive thrombocytopenia and mild neutropenia following her sixth cycle.
Two weeks after receiving cycle six of pembrolizumab, she presented to oncology clinic with weakness. Further workup revealed worsening leukopenia with white blood cell (WBC) count at 1,300/µL and absolute neutrophil count of 500/µL, platelet count of 89,000/µL, and a hemoglobin of 7.3 g/dL. Subsequent workup for pancytopenia was unrevealing with normal lactate dehydrogenase (LDH), haptoglobin, thyroid function tests, vitamin B12 and folic acid. Peripheral smear was obtained and demonstrated normocytic anemia, anisopoikilocytosis and polymorphous lymphocytes. Direct antiglobulin test and disseminated intravascular coagulation (DIC) screen were negative. She developed transfusion dependence and presented with worsening fatigue for which she was hospitalized.
She underwent a bone marrow biopsy that showed normocellular bone marrow (30%) with panhypoplasia and reactive appearing T cells with marked infiltration of cytotoxic cells in the bone marrow. Hemophagocytosis was not identified on pathologist review. CT scan of the chest, abdomen, and pelvis showed a slight increase in a previously demonstrated left inguinal mass, a minor decrease in mediastinal adenopathy, and a new 0.8 cm pulmonary nodule, however, did not meet Response Evaluation Criteria in Solid Tumors (RECIST) criteria for disease progression. A reactive process was favored over T-cell malignancy as the T cells did not show any loss of T-cell antigens, there were no definitive aggregates or marked cytologic atypia, and T-cell gene arrangement studies showed an oligoclonal pattern. Correlation with cytogenic and fluorescence in situ hybridization (FISH) studies showed normal cytogenetics by karyotype and negative FISH panel for myelodysplastic/myeloproliferative neoplasms. These findings overall favored immune-mediated bone marrow suppression as a diagnosis of exclusion.
The patient was started on steroids 1 mg/kg methylprednisolone and with gradual improvement in her cell counts, she was eventually discharged on 60 mg prednisone daily. Her hemoglobin had improved to 7.8 g/dL, with platelets at 44,000/µL and WBC at 1,300/µL at discharge.
Three days later, she was readmitted for severe anemia with hemoglobin of 5.4 g/dL along with symptoms of weakness and lightheadedness. Additionally, her WBC count had decreased to 900/µL with an absolute neutrophil count (ANC) of 400/µL. Platelets were stable at 64,000/µL. She was transfused one unit of packed red blood cells. Shortly after this transfusion, she was found to be febrile. Due to concern for a transfusion reaction, the transfusion was stopped shortly afterward.
On re-admission, prednisone was continued at 1 mg/kg. Further workup for neutropenic fever was pursued. Blood cultures were negative at 48 h. Chest X-ray demonstrated a small left pleural effusion and a few minimal patchy ground glass opacities. Laboratory results showed markedly elevated ferritin to 64,579 ng/mL, hypofibrinogenemia of 99 mg/dL, elevated triglycerides of 251 mg/dL, and elevated LDH of 1,950 U/L. All viral serologies, including Epstein-Barr virus, cytomegalovirus, parvovirus, herpes simplex virus, were negative. Fungitell and urine histoplasma antigen were negative. Given the constellation of signs and symptoms and rapid deterioration, there emerged a concern for HLH. Per the HLH-2004 trial, HLH is diagnosed based on meeting at least five of eight criteria, including fever > 38.5 °C, splenomegaly, cytopenias affecting at least two lineages, hypertriglyceridemia and/or hypofibrinogenemia, hemophagocytosis, decreased natural killer (NK)-cell activity, elevated sCD25, and hyperferritinemia [11]. This patient met modified criteria for HLH, including pancytopenia, hepatitis, fever, splenomegaly, hyperferritinemia, hypofibrinogenemia, hypertriglyceridemia, and elevated soluble interleukin (IL)-2 receptor levels at 8,645 U/mL. Furthermore, NK-cell activity tests were obtained and demonstrated profoundly low NK-cell activity with NK 50:1 of 5.3% (normal value greater than 7.8%). At the time, we also considered the possibility of sepsis. However, there was no infectious source found despite extensive imaging; blood cultures were negative, and our viral panel was unremarkable. Moreover, this patient’s extremely high ferritin level was more consistent with HLH than an acute phase reactant from sepsis. Additionally, our differential also included cytophagic histiocytic panniculitis and thrombotic thrombocytopenic purpura (TTP). This patient had no subcutaneous nodules to suggest cytophagic histiocytic panniculitis and an absence of intravascular hemolysis to suggest TTP. We favored ICI-related HLH due to constellation of symptoms and negative workup for other etiologies.
Treatment
Steroids were increased to dexamethasone 10 mg/kg. After no signs of improvement following 3 days of steroids, the patient was started on tocilizumab at 4 mg/kg as this was shown to be a potentially therapeutic option for the management of steroid refractory immune-related adverse effects via a mechanism of immune checkpoint blockade [12]. A second dose of tocilizumab at 8 mg/kg was given 48 h later with no improvement. The patient was then transitioned to etoposide at 150 mg/m2 for four doses as per HLH 1994 protocol [13].
Follow-up and outcomes
She completed treatment in the outpatient setting which she tolerated well, with improvement in her clinical symptoms and normalization of her cell lines. A PET scan with CT of the skull to mid-thigh was conducted close to discharge to restage the patient’s SCC. No new sites of avid metastatic disease were found, with some interval resolution of hypermetabolic activity associated with her thoracic lymphadenopathy and pulmonary nodules consistent with treatment response. A review of this patient’s laboratory findings may be found in Table 1; an overview of their thrombocytopenia and ferritin levels are provided in Figures 1 and 2.
 Click to view | Table 1. A Review of This Patient’s Laboratory Findings |
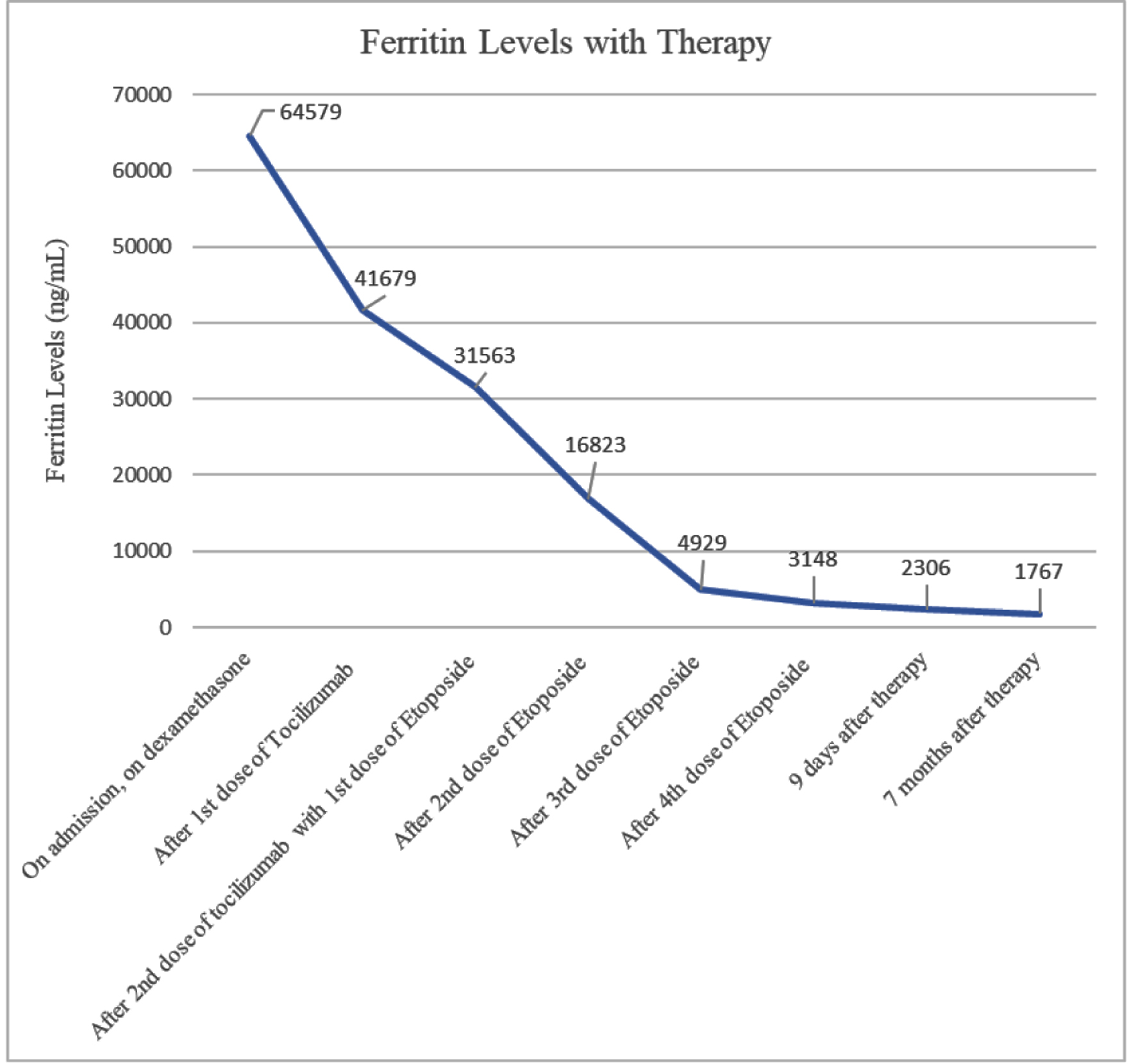 Click for large image | Figure 1. Patient’s ferritin levels over time with therapy. Patient responded quickly, but ferritin levels did not return to below normal even seven months after therapy. |
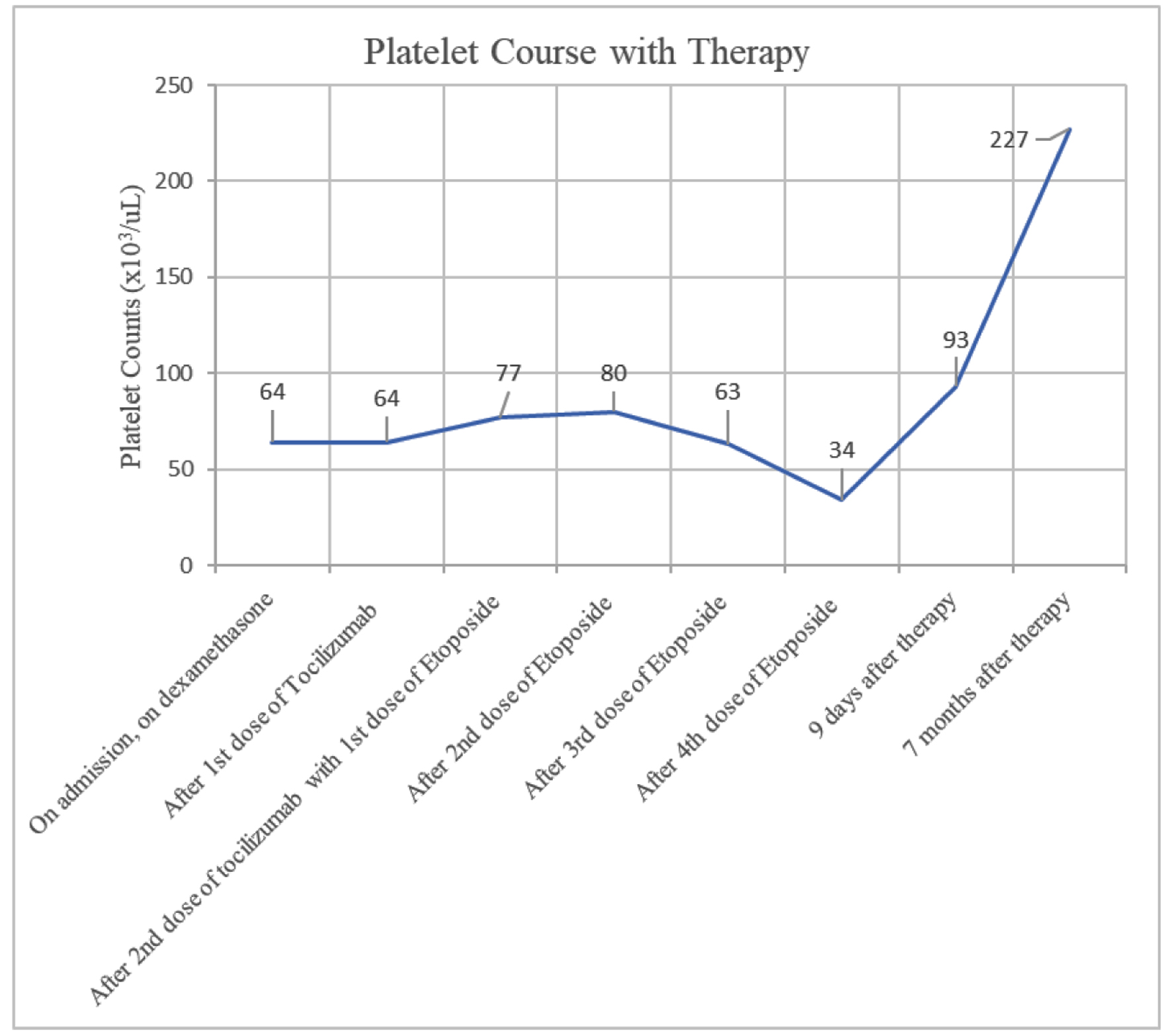 Click for large image | Figure 2. Patient’s platelet over time with therapy. Patient’s thrombocytopenia improved more gradually and only after completion of four doses of etoposide. However, thrombocytopenia did eventually resolve seven months after therapy. |
| Discussion | ▴Top |
We present an interesting case of metastatic squamous cell skin cancer with an unknown primary in a patient who developed subsequent HLH caused by pembrolizumab therapy. In a literature review, there have been reports of ICI-induced HLH in SCC of the lung, urothelial carcinoma, thymic carcinoma, Merkle cell carcinoma, breast cancer, head and neck SCC, and melanoma [2, 6, 11, 14, 15]. We contribute to the current database with this rare case of ICI-induced HLH in a patient with SCC.
There is discussion regarding aberrant T-cell overactivation as the trigger for ICI-induced HLH. Pembrolizumab acts by binding and blocking the PD-L1 receptor, preventing these receptors from interacting with PD-1-binding sites on lymphocytes. However, the PD-1 receptor is expressed on immune cells to help regulate self-tolerance, promoting the conversion of cytotoxic T cells into T-regulatory cells. Therefore, impeding this interaction may increase cytotoxic T-cell activity and lead to the development of HLH [11, 16]. Researchers have observed an inverse relationship between PD-1 expression and tumor-associated macrophage and cancer cell phagocytosis following PD-1/PD-L1 targeted therapy [14]. Moreover, there have been cases of patients treated with chimeric antigen receptor T-cell (CAR-T) therapy that have developed HLH, further demonstrating the association between T-cell activity regulation and the development of HLH [15].
HLH onset with pembrolizumab has been widely variable. Some cases report signs of HLH less than a month after starting, while others report over a year [2, 17]. As previously noted, our patient developed HLH roughly 6 months after starting pembrolizumab. Additionally, outcomes for HLH appear to vary depending on the underlying cause [18]. Recent retrospective observational cross-section studies suggest ICI-induced HLH was resolved in 61% of cases with the sole use of steroids [10, 19]. Regardless, the mortality of patients with HLH is high and one meta-analysis comprised of 26 ICI-related HLH patients found a mortality rate of 23% [16]. Patients treated with the HLH-1994 protocol have a median survival rate of 54% [20]. A higher serum ferritin or a slower rate of decline in ferritin confers a worse prognosis [21]. The prognosis is also worse in older adults, especially with those with an underlying malignancy [22]. Earlier diagnosis of HLH is expected to improve survival; thus it is critical to recognize and treat early.
Unfortunately, this disease is often difficult to diagnose. In our case, we suspected our patient was initially experiencing immune-related bone marrow suppression, when this was masking presentation of HLH. Therefore, HLH should be suspected in cases where patients on ICI develop symptoms such as cytopenia, fever, or skin rashes. As these symptoms are often vague and resemble other immune-mediated adverse events, the diagnosis is usually challenging [4].
Conclusion
We present a case report of a patient with SCC of unknown origin who developed HLH as a complication of ICI therapy. As this complication is rare, challenging, and difficult to diagnose with non-specific symptoms, practitioners should maintain a low threshold to consider HLH in patients with ICI therapy who develop cytopenias, fevers, skin rashes, or liver dysfunction. Moreover, as HLH is often life-threatening, it is crucial to intervene at the earliest.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
Informed consent has been obtained.
Author Contributions
Rosalyn Marar contributed to direct patient care during the hospitalization, literature search, and manuscript writing. Sruti Prathivadhi-Bhayankaram contributed to literature search and manuscript writing. Mridula Krishnan contributed to direct patient care during the hospitalization along with editing and supervising the entire work.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and anti-CTLA-4 therapies in cancer: mechanisms of action, efficacy, and limitations. Front Oncol. 2018;8:86.
doi pubmed - Okawa S, Kayatani H, Fujiwara K, Ozeki T, Takada K, Iwamoto Y, Minami D, et al. Pembrolizumab-induced autoimmune hemolytic anemia and hemophagocytic lymphohistiocytosis in non-small cell lung cancer. Intern Med. 2019;58(5):699-702.
doi pubmed - de La Rochefoucauld J, Noel N, Lambotte O. Management of immune-related adverse events associated with immune checkpoint inhibitors in cancer patients: a patient-centred approach. Intern Emerg Med. 2020;15(4):587-598.
doi pubmed - Takahashi H, Koiwa T, Fujita A, Suzuki T, Tagashira A, Iwasaki Y. A case of pembrolizumab-induced hemophagocytic lymphohistiocytosis successfully treated with pulse glucocorticoid therapy. Respir Med Case Rep. 2020;30:101097.
doi pubmed - Kennedy LB, Salama AKS. A review of cancer immunotherapy toxicity. CA Cancer J Clin. 2020;70(2):86-104.
doi pubmed - Sadaat M, Jang S. Hemophagocytic lymphohistiocytosis with immunotherapy: brief review and case report. J Immunother Cancer. 2018;6(1):49.
doi pubmed - Parikh SA, Kapoor P, Letendre L, Kumar S, Wolanskyj AP. Prognostic factors and outcomes of adults with hemophagocytic lymphohistiocytosis. Mayo Clin Proc. 2014;89(4):484-492.
doi pubmed - Daver N, McClain K, Allen CE, Parikh SA, Otrock Z, Rojas-Hernandez C, Blechacz B, et al. A consensus review on malignancy-associated hemophagocytic lymphohistiocytosis in adults. Cancer. 2017;123(17):3229-3240.
doi pubmed - Dupre A, Michot JM, Schoeffler A, Frumholtz L, Baroudjian B, Delyon J, Lebbe C, et al. Haemophagocytic lymphohistiocytosis associated with immune checkpoint inhibitors: a descriptive case study and literature review. Br J Haematol. 2020;189(5):985-992.
doi pubmed - Noseda R, Bertoli R, Muller L, Ceschi A. Haemophagocytic lymphohistiocytosis in patients treated with immune checkpoint inhibitors: analysis of WHO global database of individual case safety reports. J Immunother Cancer. 2019;7(1):117.
doi pubmed - Kalmuk J, Puchalla J, Feng G, Giri A, Kaczmar J. Pembrolizumab-induced Hemophagocytic Lymphohistiocytosis: an immunotherapeutic challenge. Cancers Head Neck. 2020;5:3.
doi pubmed - Stroud CR, Hegde A, Cherry C, Naqash AR, Sharma N, Addepalli S, Cherukuri S, et al. Tocilizumab for the management of immune mediated adverse events secondary to PD-1 blockade. J Oncol Pharm Pract. 2019;25(3):551-557.
doi pubmed - Henter JI, Samuelsson-Horne A, Arico M, Egeler RM, Elinder G, Filipovich AH, Gadner H, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100(7):2367-2373.
doi pubmed - Hantel A, Gabster B, Cheng JX, Golomb H, Gajewski TF. Severe hemophagocytic lymphohistiocytosis in a melanoma patient treated with ipilimumab + nivolumab. J Immunother Cancer. 2018;6(1):73.
doi pubmed - Al-Samkari H, Snyder GD, Nikiforow S, Tolaney SM, Freedman RA, Losman JA. Haemophagocytic lymphohistiocytosis complicating pembrolizumab treatment for metastatic breast cancer in a patient with the PRF1A91V gene polymorphism. J Med Genet. 2019;56(1):39-42.
doi pubmed - Davis EJ, Salem JE, Young A, Green JR, Ferrell PB, Ancell KK, Lebrun-Vignes B, et al. Hematologic complications of immune checkpoint inhibitors. Oncologist. 2019;24(5):584-588.
doi pubmed - Laderian B, Koehn K, Holman C, Lyckholm L, Furqan M. Association of hemophagocytic lymphohistiocytosis and programmed death 1 checkpoint inhibitors. J Thorac Oncol. 2019;14(4):e77-e78.
doi pubmed - Hayden A, Park S, Giustini D, Lee AY, Chen LY. Hemophagocytic syndromes (HPSs) including hemophagocytic lymphohistiocytosis (HLH) in adults: A systematic scoping review. Blood Rev. 2016;30(6):411-420.
doi pubmed - Dufranc E, Del Bello A, Belliere J, Kamar N, Faguer S, TAIDI (Toulouse Acquired Immune Deficiency and Infection) study group. IL6-R blocking with tocilizumab in critically ill patients with hemophagocytic syndrome. Crit Care. 2020;24(1):166.
doi pubmed - Trottestam H, Horne A, Arico M, Egeler RM, Filipovich AH, Gadner H, Imashuku S, et al. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood. 2011;118(17):4577-4584.
doi pubmed - Lin TF, Ferlic-Stark LL, Allen CE, Kozinetz CA, McClain KL. Rate of decline of ferritin in patients with hemophagocytic lymphohistiocytosis as a prognostic variable for mortality. Pediatr Blood Cancer. 2011;56(1):154-155.
doi pubmed - Arca M, Fardet L, Galicier L, Riviere S, Marzac C, Aumont C, Lambotte O, et al. Prognostic factors of early death in a cohort of 162 adult haemophagocytic syndrome: impact of triggering disease and early treatment with etoposide. Br J Haematol. 2015;168(1):63-68.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.