

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://www.thejh.org |
Case Report
Volume 11, Number 6, December 2022, pages 233-239

Bone Involvement as a Primary Rare Manifestation of Waldenstrom Macroglobulinemia: A Case Report and Prevalence in a Nationwide Population-Based Cohort Study
Khazra Bhattia, Aqsa Nazira, Simon Ostergaardb, Lone Schejbelc, Peter Norgaardc, Lise M.R. Gjerdrumb, d, Mahnaz Moghaddase, Torsten H. Nielsenf, Lars Munksgaardg, Lars M. Pedersend, g, h
aInstitute of Science and Environment, Roskilde University, Roskilde, Denmark
bDepartment of Pathology, Zealand University Hospital, Roskilde, Denmark
cDepartment of Pathology, Herlev University Hospital, Herlev, Denmark
dDepartment of Clinical Medicine, University of Copenhagen, Copenhagen, Denmark
eDepartment of Radiology, Herlev University Hospital, Herlev, Denmark
fDepartment of Hematology, Copenhagen University Hospital, Copenhagen, Denmark
gDepartment of Hematology, Zealand University Hospital, Roskilde, Denmark
hCorresponding Author: Lars M. Pedersen, Department of Hematology, Zealand University Hospital, DK-4000 Roskilde, Denmark
Manuscript submitted October 31, 2022, accepted December 13, 2022, published online December 30, 2022
Short title: Bone Involvement in WM
doi: https://doi.org/10.14740/jh1073
| Abstract | ▴Top |
Bone involvement is a rare extranodal manifestation in patients with malignant lymphoproliferative diseases and has also been noted as a rare event in patients with Waldenstrom macroglobulinemia (WM). However, the actual prevalence has not been previously reported. We describe an unusual case of a patient with WM who presented with lower back pain and focal bone lesions at initial diagnosis. Magnetic resonance imaging (MRI) revealed multiple vertebral fractures. Positron emission tomography (PET) detected only nodal changes without pathological skeletal-related metabolic activity. Lymph node and bone marrow biopsies combined with an immunoglobulin M (IgM) M component revealed the diagnosis of WM. A next-generation sequencing (NGS) analysis using a targeted lymphoma panel of 59 recurrently mutated genes in lymphoid neoplasms showed mutations in the MYD88 and CD79B genes. After treatment with rituximab and bendamustine, the patient achieved a partial remission and pain relief. After 3 years of stable disease, a spontaneous subcapital fracture at the base of the femoral neck and new vertebral compression fractures occurred. Whole-body low-dose computed tomography (WB-LDCT) and bone density (dual energy X-ray absorptiometry (DEXA)) scan revealed marked osteopenia. After insertion of a hip prosthesis, examination of the removed hip showed infiltration of clonal lymphoplasmacytic cells. Our case confirms that one must be aware that bone involvement in patients with WM can occur as a rare manifestation. Interestingly, the MYD88/CD79B-mutated (MCD) genotype in diffuse large B-cell lymphoma is characterized by extranodal involvement and may also be involved in the pathogenesis of skeletal-related disease in the present case. As a follow-up to this unusual case, we have carried out an analysis based on the Danish Lymphoma Registry (LYFO) covering the entire national population in the period 2000 - 2020. The registry study included a cohort of 2,459 patients with WM and lymphoplasmacytic lymphoma. Our data revealed that primary bone involvement at diagnosis occurs in 1.75% of adults with WM. To the best of our knowledge, this is the first report of the prevalence of skeletal-related disease in a large nationwide cohort and defines bone involvement as an exceedingly rare event in WM.
Keywords: Waldenstrom macroglobulinemia; Lymphoplasmacytic lymphoma; B-cell lymphoma; Bone involvement; Bone lesions; Extramedullary disease
| Introduction | ▴Top |
Waldenstrom macroglobulinemia (WM) is a distinct indolent B-cell lymphoproliferative disorder characterized by lymphoplasmacytic lymphoma (LPL) in the bone marrow (BM) and an immunoglobulin M (IgM) monoclonal gammopathy [1]. The diagnosis is based on identification of LPL cells in the BM with an immunophenotypic profile characterized by a heterogeneous population of post-germinal center, mature B-cells, ranging from small B-lymphocytes (surface IgM, CD19, CD20, CD22 dim, and other B-cell markers) to a plasmacytic component of fully differentiated plasma cells (CD138+). Immunostaining with Ki-67 typically shows a low proliferation rate. Molecular hallmarks of WM are somatic mutations in MYD88 (> 95%) and CXCR4 (> 30%) genes [2-4]. MYD88 mutation is consistent with but not specific for WM/LPL [5].
Most patients with WM show an indolent clinical course and extramedullary involvement is a rare finding. Based on individual case reports, symptomatic focal lytic bone lesions in WM/LPL are a known, albeit rare, complication of WM [6-11]. However, the actual prevalence has never been reported. The skeletal involvement is associated with a predominance of plasmacytic morphology in some case reports [12, 13]. The clinical presentation of lytic bone lesions in WM both raises the suspicion of a high-grade transformation or multiple myeloma (MM). Molecular characteristics of WM with this rare manifestation remain unknown. Thus, MYD88 mutational status and other key molecular findings are only available in very few reports and the diagnosis may have been associated with some uncertainty including IgM MM as a differential diagnosis.
In the current report, we present a rare case of WM with bone lesions as primary manifestation of the disease. The case is illustrated by clinical findings, imaging, and molecular characteristics. We also present real-life data from a national population-based survey, which to the best of our knowledge, is the first report on the prevalence of bone lesions in a large cohort of newly diagnosed WM/LPL patients.
| Case Report | ▴Top |
Investigations, diagnosis, and treatment
A 65-year-old Caucasian male patient presented with complaints of increasing low back pain over a year. Fractures of the Th7, Th11, Th12, L1, and L3 vertebrae were demonstrated by a magnetic resonance imaging (MRI) scan (Fig. 1a). The patient had no other medical conditions and received no medications predisposing to osteoporosis. A computed tomography (CT) scan revealed enlarged lymph nodes both in the thoracic and abdominal region. A biopsy from a right inguinal lymph node was performed showing infiltration of small, uniform B-lymphocytes and plasmacytoid cells with strong expression of CD20, PAX5, and BCL2 (Fig. 2a, b). The lymphocytes displayed a diffuse infiltration pattern and did not express cyclin D1, CD5, BCL6, or CD10. Scattered polytypic plasma cells and an increased number of mast cells were noted. A polymerase chain reaction (PCR) analysis demonstrated a MYD88 L265P mutation. The WM diagnosis was established by a BM biopsy revealing infiltration of monoclonal LPL cells (Fig. 2c, d) and flow cytometry with 17% monoclonal kappa B-cells with a nonspecific phenotype compatible with LPL (Fig. 2e). There was no sign of amyloid deposition. Additionally, we performed next-generation sequencing (NGS) analysis using our in-house targeted lymphoma panel of 59 recurrently mutated genes of known diagnostic, prognostic, or therapeutic significance in lymphoid neoplasms. It is based on Ion Torrent Ampliseq on demand assays for 51 genes and Ion Torrent Ampliseq custom design assays for eight genes. The analysis covers SNV, MNV and INDELs of all exons and consensus splice sites. Sequencing was done on the Ion Torrent S5 XL system using manual library preparation and the Ion Chef system as described by the manufacturer (Thermo Fisher Scientific). Variants with a variant allele frequency < 5% were not reported. In the present case, MYD88 (c.818T>C, p.Leu273Pro, 70%) and CD79B (c.589T>C, p.Tyr197His, 40%; c.582T>G, p.Asp194Glu, 40%) mutations were detected while CXCR4, ARID1A, and TP53 were found to be wild type. The frequently mutated genes in multiple myeloma covered by the panel (KRAS, NRAS, BRAF, TP53, IR4, and ATM) were not mutated.
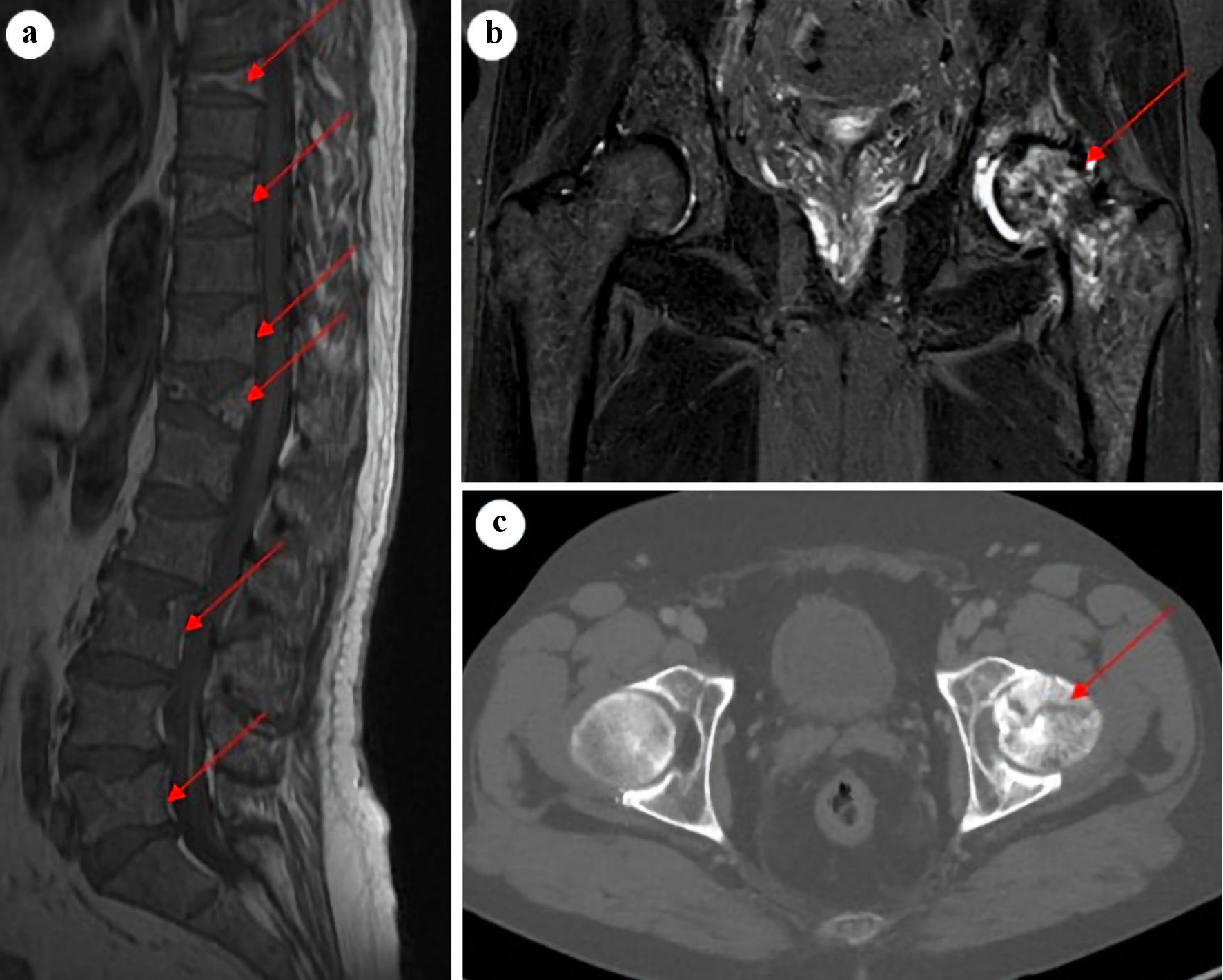 Click for large image | Figure 1. Sagittal T1-weighted MRI of lumbar and thoracic spine (a) showing multiple vertebral fractures and osteopenia (arrows). Coronal STIR MRI (b) and axial CT image (c) of the hip joints with a suspected malignant process in the left proximal femur and a subcapital fracture (arrows), with accompanying synovitis and accumulation of fluid in the left hip joint. MRI: magnetic resonance imaging; CT: computed tomography. |
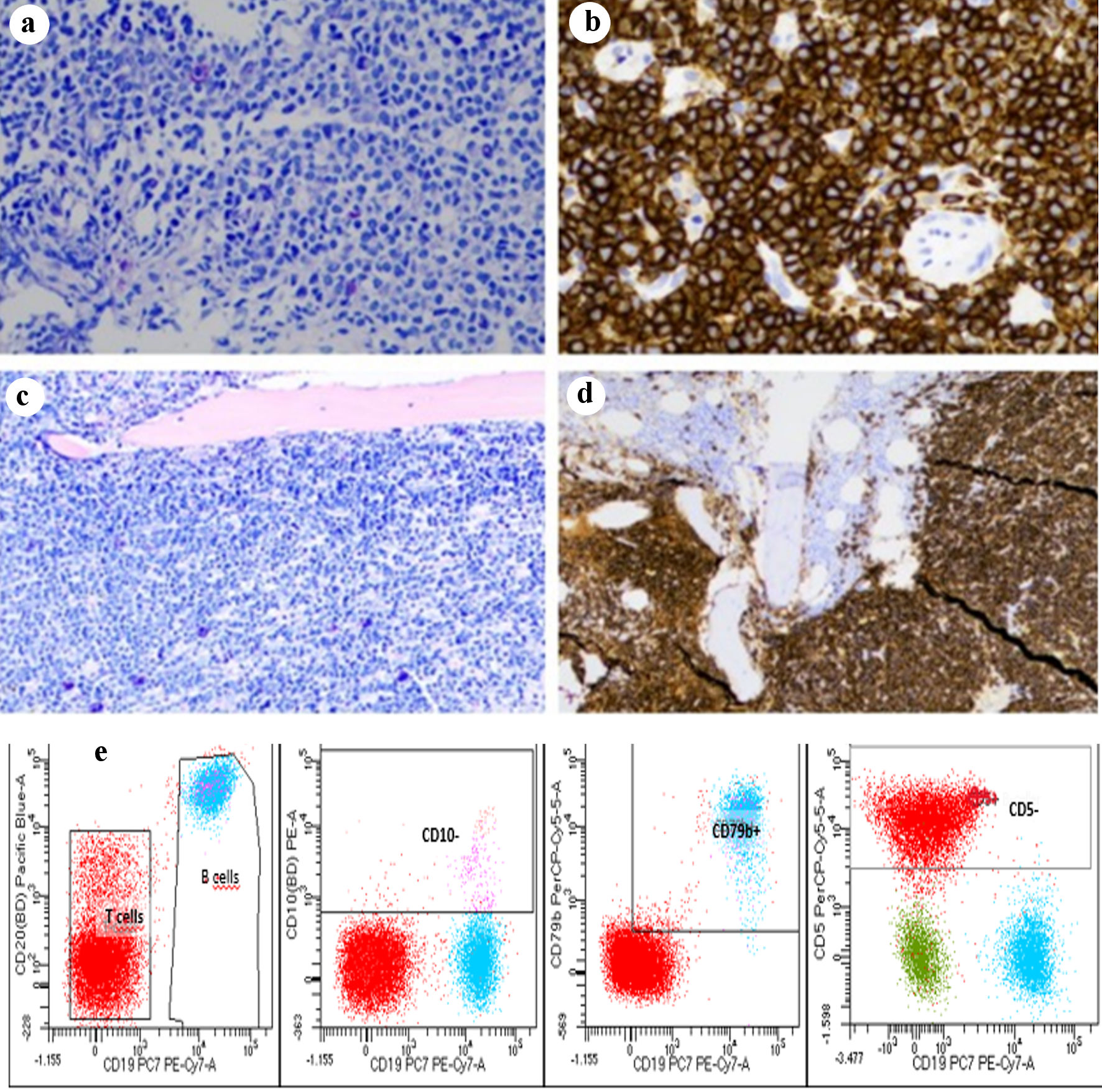 Click for large image | Figure 2. Lymph node biopsy with diffuse infiltration of small, uniform B-lymphocytes and plasmacytoid cells (a) with strong expression of CD20 (b). Bone marrow infiltration of monoclonal LPL cells (c) expressing CD20 (d). Flow cytometry identifies a distinct clonal population of B cells (CD19+, CD20+) which are negative for CD5 and CD10 (e). LPL: lymphoplasmacytic lymphoma. |
The patient was characterized as intermediate risk according to the revised-International Prognostic Scoring System for WM (rIPSSWM) score [14]. The IgM kappa M-component was 6 g/L, β2-microglobulin 6.4 mg/L, and serum albumin 24 g/L at diagnosis. Hemoglobin, platelet counts, and lactate dehydrogenase (LDH) were normal. There was no evidence of hypercalcemia. A supplementary 18F-fluorodeoxyglucose (18F-FDG) positron emission tomography (PET) scan demonstrated a few enlarged lymph nodes with a metabolic activity equivalent to Deauville 3 - 4 but no osseous structures showed pathological metabolic activity (Deauville 1 - 2). The patient was treated with six courses of rituximab (375 mg/m2) and bendamustine (90 mg/m2) and obtained a partial response with ≥ 50% reduction of the serum monoclonal IgM spike from 6 to 2 g/L and a ≥ 50% reduction of adenopathy. The patient experienced pain relief following treatment suggesting a causal relationship between the presence of malignant cells, bone destruction, and pain.
Three years after initial diagnosis, the patient complained of persistent pain in the left hip region and a MRI was conducted. An inhomogeneous bone structure with suspected malignancy proximally in the left femur and a subcapital fracture at the base of the femoral neck was noted (Fig. 1). Bone density (dual energy X-ray absorptiometry (DEXA)) scan revealed marked osteopenia in both hips and the spine with a bone mineral density (BMD) T-score of -1.5 and -2.7, respectively. The patient underwent a surgical procedure with insertion of a hip prosthesis followed by consolidating radiotherapy (5 Gy × 5). The removed hip showed nodular infiltration consisting of a mixture of plasmacytoid cells and small lymphocytes in accordance with the LPL diagnosis without any signs of transformation to aggressive lymphoma.
Follow-up and outcomes
Until the new bone-related events with pathologic fractures described above, the patient had shown stable remission without pain or symptoms related to the disease. Treatment with bisphosphonate was initiated and at follow-up 1 year after the surgical procedure, the patient had no bone-related symptoms. During this period, the patient was followed with regular whole-body low-dose computed tomography (WB-LDCT) and DEXA scans, which demonstrated no further bone-related events or worsening of the osteopenia. However, the IgM M-component was slowly increasing from 2 to 6 g/L, and the patient experienced symptoms suggestive of a peripheral neuropathy. Neurophysiological examinations confirmed a mixed demyelinating and axonal nerve damage. Examination of the cerebrospinal fluid (CSF) revealed no malignant monoclonal cells. Polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, skin changes (POEMS) syndrome was considered but the patient did not fulfill the diagnostic criteria and level of serum vascular endothelial growth factor (VEGF) was normal.
Real-life data from the Danish Lymphoma Registry
Due to the lack of data in the literature, we have investigated the prevalence of bone involvement in WM/LPL in a large nationwide population-based cohort. Patients (diagnosis code World Health Organization (WHO)/International Classification of Diseases 10th Revision (ICD-10) 9671) were identified through a prescreening process in the Danish Lymphoma Registry (LYFO) [15]. The registry study was approved by the Danish National Committee on Health Research Ethics (approval number 2113049). The LYFO database has been shown to have a coverage of 95% of all Danish patients with lymphoma in a recent quality assessment [15]. Clinical data for newly diagnosed WM/LPL in the period 2000 - 2020 were extracted from the LYFO registry. Data included diagnosis, age, gender, bone involvement, other extramedullary sites involved, laboratory findings, and treatment vs. no treatment at primary diagnosis. The presence of bone lesions was based on a clinical assessment including the results of imaging. Guidelines in Denmark recommend a CT scan for the primary investigation at diagnosis. However, data for the use of imaging modalities are not available in the registry.
In total, the study cohort comprised 2,459 patients diagnosed between January 2000 and December 2020. The number of patients diagnosed as WM and LPL were 1,977 (80.4%) and 482 (19.6%), respectively. Data on skeletal involvement were not available in five patients. Therefore, a study population of 2,454 patients was included in the analyses of which the number of patients with bone involvement at diagnosis was 43 (1.75%). Among patients with bone involvement, extramedullary manifestations other than bone lesions (lung, liver, and central nervous system) were registered in three (7%) of the patients whereas 40 (93%) patients had bone lesions as the only registered extranodal site. Clinical data comparing patients with and without involvement of bones are outlined in Table 1. Patients with bone involvement were characterized by a significantly higher proportion of patients with a LPL diagnosis, presence of B symptoms and need for treatment (Table 1).
 Click to view | Table 1. Clinical Data Characteristics of 2,454 Newly Diagnosed WM/LPL Patients With and Without Bone Involvement |
| Discussion | ▴Top |
Based on available reports, cortical and trabecular involvement of bones and generalized osteopenia must be considered as a rare manifestation of WM/LPL [6-11]. Lytic bone lesions have previously been reported as a first presenting feature at the diagnosis of WM [12, 13]. However, the occurrence of bone involvement in WM/LPL has so far only been described in selected smaller case series and therefore the actual prevalence is unknown. We report the real-life prevalence in a large nationwide population of patients with newly diagnosed WM/LPL over the past two decades. We have shown a very low frequency of patients (1.75%) presenting with clinical findings at diagnosis suggesting the presence of bone lesions. As expected, the need for systemic treatment appears to be higher for patients with bone involvement. Use of imaging modalities was not recorded, and it is assumed that identification of lytic bone lesions could be underestimated because of a lack of diagnostic vigilance and varying use of imaging in relation to pathological bone lesions. It also must be considered that our data only include patients at the time of diagnosis and the presence of bone manifestations may have increased if data also included events for the entire course of the disease. Thus, in previous studies, it has been reported that bone involvement can occur both early and late in the course of WM/LPL [16, 17].
Extramedullary presentation in WM/LPL is a rare manifestation only reported in a few studies. In a large single-center study of WM patients, extramedullary involvement was generally associated with poor prognosis [18]. Treatment with chemotherapy or targeted therapy appeared to be less effective in patients with extramedullary disease but no cases of skeletal involvement were reported. Another single-institution survey revealed a frequency of 4.4% having extramedullary involvement (lymph nodes, liver, and spleen not included) [19]. Extramedullary sites most involved were pulmonary (30%), soft tissue (21%), CSF (23%), renal (8%), and bone (9%). This study therefore supports that the occurrence of bone involvement is extremely rare in WM also in patients with extramedullary disease.
Our case report illustrates the importance of diagnostic awareness in a patient with unexplained pain complaints. When the patient was thoroughly examined, potentially malignant bone lesions were revealed. Guidelines for the use of diagnostic imaging modalities to identify bone involvement in WM/LPL are lacking. In a retrospective study of highly selected patients, PET-CT and MRI demonstrated bone disease in 24% and 17% of the cases, respectively [10]. In our patient, the areas of bone involvement were not FDG-avid. Interestingly, there seems to be no clear correlation between general disease activity and bone involvement in the case of our patient. The patient presented with marked signs of bone destruction and osteopenia despite a clinical indolent WM. Focal bone lesions without cortical involvement may also cause pain. In a case series of six patients without cortical or trabecular bone involvement, painful focal areas with abnormal signal within the medulla were demonstrated on MRI [16, 17]. These foci with significantly higher lymphoid infiltration (> 80%) were not identified on CT and only mildly FDG-avid. In our case with cortical destruction, the biopsy from the left hip showed clear infiltration of small lymphocytes with LPL phenotype and plasmacytoid B cells, but infiltration rate could not be reliably assessed in the available material.
Several genes are recurrently mutated in malignant lymphoproliferative diseases. In LPL/WM, NGS has identified common recurrent somatic mutations in the MYD88 (> 90%), CXCR4 (> 30%), ARID1A (17%), and CD79B (8-15%) genes [20]. The mutational landscape has been shown to influence the clinical phenotype and outcome. Patients with MYD88 and ARID1A mutations show greater BM disease involvement, and lower hemoglobin and platelet count. Asymptomatic WM patients harboring a CXCR4 mutation have a shorter treatment-free survival than patients with wild-type CXCR4 [21]. CXCR4 mutations also promote disease progression and resistance to ibrutinib treatment [22]. The C1013G/CXCR4 variant appears to be associated with an increased risk of dissemination to extramedullary organs [4]. CD79A and CD79B mutations can be found in approximately 10% of WM patients [23]. However, the connection between bone disease in WM/LPL and genetic alterations has not been clarified. In our patient, we demonstrated coexistence between MYD88 and CD79B mutations and the patient had wild-type CXCR4. CD79B has also been associated with diffuse large B-cell lymphoma (DLBCL) as shown in a study with contributions from our group [24]. Thus, MYD88/CD79B-mutated (MCD) genotype is a genetic subgroup occurring in around 8% of DLBCLs [25]. The MCD genotype is characterized by poor prognosis and extranodal involvement. CD79B is a component of the B-cell receptor (BCR) with a role in chronic-active BCR and NF-κB signaling pathway. Hypothetically, the cooccurrence of MYD88 and CD79B mutations may be involved in the pathogenesis of extramedullary disease in WM and a harbinger of transformation to DLBCL. Further studies on a larger cohort of patients are needed to elucidate the pathophysiological mechanisms behind bone disease in WM.
Molecular findings may also support the prediction of therapeutic response. Thus, CD79B upregulation in ABC-type DLBCL is associated with resistance to inhibition of BCR signaling with the Bruton tyrosine kinase (BTK) inhibitor ibrutinib [26]. MYD88-mutated and CXCR4 wild-type patients have also demonstrated a longer response to ibrutinib in WM [27]. However, the optimal treatment strategy for bone disease in WM/LPL is unknown. Our patient was treated with rituximab and bendamustine with a marked initial response to both pain, lymphadenopathy, and M component. Assessed on imaging studies and DEXA scan, bone involvement remained unchanged and stable. Others have reported similar efficacy using immunochemotherapy in patients with bone involvement [8]. A favorable response on lytic lesions with the BTK inhibitor ibrutinib has also been reported [28, 29]. Data on the efficacy of bisphosphonates are lacking. Focal lytic lesions may also respond to radiotherapy [30]. Further research is warranted on the ability of novel agents to reverse bone turnover in WM/LPL patients as well as on the utility of anti-resorptive agents in patients with lytic bone disease.
Learning points
Bone lesions in WM/LPL are a rare but well-described and heterogenous manifestation of WM/LPL. We describe an unusual case with primary bone involvement at diagnosis and in the BM biopsy presence of somatic mutations in the MYD88 and CD79B genes. We also present real-life data for the frequency of bone manifestations in a large nationwide cohort covering two decades. Despite its rarity, it is recommended that evaluation of patients with newly diagnosed WM/LPL considers possible involvement of bones, not least in patients with unexplained pain. Further studies are needed to explore the molecular characteristics related to the bone manifestations. Furthermore, there is a lack of knowledge about the prognostic impact of bone involvement in WM and the optimal therapeutic strategy for these patients.
Acknowledgments
We wish to thank our patient and his family for participating in this study.
Financial Disclosure
None to declare.
Conflict of Interest
The authors have no conflict of interest to declare.
Informed Consent
Informed consent was obtained from the patient.
Author Contributions
THN, LM, and LMP participated in the clinical care of the patient. KB, AN, and LMP wrote the manuscript. SO provided data from the Danish National Lymphoma Registry; MM and LMRG created the accompanying figures. All authors contributed to the editing of the manuscript. All authors read and approved the final manuscript.
Data Availability
The authors declare that data supporting the findings of this study are available within the article. Any questions regarding data availability should be directed to the corresponding author.
Abbreviations
LPL: lymphoplasmacytic lymphoma; WM: Waldenstrom macroglobulinemia; MRI: magnetic resonance imaging; CT: computed tomography; PET: positron emission tomography; NGS: next-generation sequencing; WB-LDT: whole-body low-dose computed tomography; DEXA: dual energy X-ray absorptiometry; rIPSSWM: revised-International Prognostic Scoring System for WM; BMD: bone mineral density; VEGF: vascular endothelial growth factor; BM: bone marrow
| References | ▴Top |
- WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Revised 4th ed. Geneva: WHO Press; 2017.
- Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao Y, Sheehy P, et al. MYD88 L265P somatic mutation in Waldenstrom's macroglobulinemia. N Engl J Med. 2012;367(9):826-833.
doi pubmed - Varettoni M, Arcaini L, Zibellini S, Boveri E, Rattotti S, Riboni R, Corso A, et al. Prevalence and clinical significance of the MYD88 (L265P) somatic mutation in Waldenstrom's macroglobulinemia and related lymphoid neoplasms. Blood. 2013;121(13):2522-2528.
doi pubmed - Hunter ZR, Xu L, Yang G, Zhou Y, Liu X, Cao Y, Manning RJ, et al. The genomic landscape of Waldenstrom macroglobulinemia is characterized by highly recurring MYD88 and WHIM-like CXCR4 mutations, and small somatic deletions associated with B-cell lymphomagenesis. Blood. 2014;123(11):1637-1646.
doi pubmed - Jimenez C, Sebastian E, Chillon MC, Giraldo P, Mariano Hernandez J, Escalante F, Gonzalez-Lopez TJ, et al. MYD88 L265P is a marker highly characteristic of, but not restricted to, Waldenstrom's macroglobulinemia. Leukemia. 2013;27(8):1722-1728.
doi pubmed - Koehler M, Moita F, Cabecadas J, Gomes da Silva M. Mixed lytic and blastic bone lesions as a presenting feature of Waldenstrom macroglobulinemia: case report and review of the literature. Clin Lymphoma Myeloma Leuk. 2020;20(2):e87-e91.
doi pubmed - Lin JM, Yuan XJ, Zhang L, Li G, Gan XR, Xu WH. Does Waldenstrom's macroglobulinemia also cause bone destruction? A rare case report. J Int Med Res. 2022;50(4):1-8.
doi pubmed - Baksh M, Jiang L, Bhatia U, Alegria V, Sher T, Roy V, Chanan-Khan A, et al. Management of lytic bone disease in lymphoplasmacytic lymphoma: A case report and review of the literature. Clin Case Rep. 2021;9(12):e05181.
doi pubmed - Schlesinger N, Neustadter L, Schumacher HR. Lytic bone lesions as a prominent feature in Waldenstrom's macroglobulinemia. J Clin Rheumatol. 2000;6(3):150-153.
doi pubmed - Papanikolaou X, Waheed S, Barlogie B, Alapat DV, Yoon D, Kumar M, Matin A, et al. Waldenstrom's macroglobulinemia associated bone disease the UAMS experience. Blood. 2014;124(21):2999.
doi - Mehmood K, Naqvi IH, Shah SR, Zakir N, Ali SM. Waldenstroms macroglobulinemia patient presenting with rare 'lytic' lesions and hypercalcemia: a diagnostic dilemma. J Clin Diagn Res. 2014;8(11):FD10-11.
doi pubmed - Marks MA, Tow DE, Jay M. Bone scanning in Waldenstrom's macroglobulinemia. J Nucl Med. 1985;26(12):1412-1414.
- Pujani M, Kushwaha S, SethiN, Beniwal A, Shukla S. Waldenstrom's macroglobulinemia presenting with lytic bone lesions: a rare presentation. Blood Res. 2013;48(3):226-234.
doi pubmed - Kastritis E, Morel P, Duhamel A, Gavriatopoulou M, Kyrtsonis MC, Durot E, Symeonidis A, et al. A revised international prognostic score system for Waldenstrom's macroglobulinemia. Leukemia. 2019;33(11):2654-2661.
doi pubmed - Arboe B, El-Galaly TC, Clausen MR, Munksgaard PS, Stoltenberg D, Nygaard MK, Klausen TW, et al. The Danish National Lymphoma Registry: coverage and data quality. PLoS One. 2016;11(6):e0157999.
doi pubmed - Arulogun S, Abbasi MA, Pomplun S, O'Neill AT, Wan S, Kayani I, D'Sa S. Focal, symptomatic bone marrow lesions in Waldenstrom macroglobulinaemia characterised by dense infiltration with lymphoplasmacytic lymphoma: A newly defined indication for systemic treatment. Blood. 2020;136(Suppl 1):3-4.
doi - Arulogun SO, Abbasi MA, Pomplun S, O'Neill AT, Wan S, Wechalekar A, D'Sa SP. Clinicoradiopathological correlation of symptomatic focal bone marrow lesions in Waldenstrom Macroglobulinaemia. Leuk Lymphoma. 2022;63(6):1496-1499.
doi pubmed - Cao X, Ye Q, Orlowski RZ, Wang X, Loghavi S, Tu M, Thomas SK, et al. Waldenstrom macroglobulinemia with extramedullary involvement at initial diagnosis portends a poorer prognosis. J Hematol Oncol. 2015;8:74.
doi pubmed - Banwait R, Aljawai Y, Cappuccio J, McDiarmid S, Morgan EA, Leblebjian H, Roccaro AM, et al. Extramedullary Waldenstrom macroglobulinemia. Am J Hematol. 2015;90(2):100-104.
doi pubmed - Treon SP, Xu L, Guerrera ML, Jimenez C, Hunter ZR, Liu X, Demos M, et al. Genomic landscape of Waldenstrom macroglobulinemia and its impact on treatment strategies. J Clin Oncol. 2020;38(11):1198-1208.
doi pubmed - Varettoni M, Zibellini S, Defrancesco I, Ferretti VV, Rizzo E, Malcovati L, Galli A, et al. Pattern of somatic mutations in patients with Waldenstrom macroglobulinemia or IgM monoclonal gammopathy of undetermined significance. Haematologica. 2017;102(12):2077-2085.
doi pubmed - Cao Y, Hunter ZR, Liu X, Xu L, Yang G, Chen J, Patterson CJ, et al. The WHIM-like CXCR4(S338X) somatic mutation activates AKT and ERK, and promotes resistance to ibrutinib and other agents used in the treatment of Waldenstrom's Macroglobulinemia. Leukemia. 2015;29(1):169-176.
doi pubmed - Hunter ZR, Yang G, Xu L, Liu X, Castillo JJ, Treon SP. Genomics, signaling, and treatment of Waldenstrom macroglobulinemia. J Clin Oncol. 2017;35(9):994-1001.
doi pubmed - Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, Leppa S, et al. Genetic and functional drivers of diffuse large B cell lymphoma. Cell. 2017;171(2):481-494.e415.
doi pubmed - Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, Roulland S, et al. Genetics and pathogenesis of diffuse large B-cell lymphoma. N Engl J Med. 2018;378(15):1396-1407.
doi pubmed - Kim JH, Kim WS, Ryu K, Kim SJ, Park C. CD79B limits response of diffuse large B cell lymphoma to ibrutinib. Leuk Lymphoma. 2016;57(6):1413-1422.
doi pubmed - Treon SP, Tripsas CK, Meid K, Warren D, Varma G, Green R, Argyropoulos KV, et al. Ibrutinib in previously treated Waldenstrom's macroglobulinemia. N Engl J Med. 2015;372(15):1430-1440.
doi pubmed - Shinohara M, Chang BY, Buggy JJ, Nagai Y, Kodama T, Asahara H, Takayanagi H. The orally available Btk inhibitor ibrutinib (PCI-32765) protects against osteoclast-mediated bone loss. Bone. 2014;60:8-15.
doi pubmed - Tucker DL, Mihailescu L, Riordan R, Rule S. Remineralization of lytic bone disease in a patient with small lymphocytic lymphoma using ibrutinib. Br J Haematol. 2017;178(1):153-155.
doi pubmed - Priyanka P, Mercier R, Raiker R, Potugari B. Distal tibia and foot involvement in a patient with Waldenstrom's macroglobulinemia. WMJ. 2018;117(2):88-91.
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.