

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://www.thejh.org |
Letter to the Editor
Volume 12, Number 4, August 2023, pages 197-200

Hemolytic Anemia Requiring Splenectomy in Leigh-Like Syndrome due to the Variant m.10191T>C in MT-ND3
Shaundra M. Newsteada ![]() , Josef Finstererb, c
, Josef Finstererb, c ![]()
aHeatSync Biochemistry Laboratory, Mesa, AZ 85201, USA
bNeurology and Neurophysiology Center, Vienna, Austria
cCorresponding Author: Josef Finsterer, Neurology and Neurophysiology Center, Vienna, Austria
Manuscript submitted April 4, 2023, accepted May 12, 2023, published online July 12, 2023
Short title: LLS Manifesting With Hemolytic Anemia
doi: https://doi.org/10.14740/jh1122
| To the Editor | ▴Top |
Leigh syndrome is a syndromic mitochondrial disorder (MID), most commonly and clinically characterized by early-onset cognitive impairment, developmental delay, seizures, hypotonia, nutritional problems, and symmetric changes in the basal ganglia and brainstem. The effects of organs other than the brain, such as the heart, intestines, endocrine system, or blood cells, have only rarely been reported. Leigh syndrome is mainly congenital in children and rarely occurs in adults. Hemolytic anemias are a heterogeneous group of hematologic disorders in which red blood cells (RBCs) are destroyed in either an extravascular or intravascular manner [1]. One cause of hemolytic anemia is poikilocytosis, which describes the state of the RBC bimembranes no longer being a biconcave disc shape, but instead, any type of shape [2]. When this occurs, phosphatidylserine from the inner membrane is externally exposed, and the blood cell is marked for destruction by the complement system or sequestered by splenic macrophages [3]. This case of a patient with Leigh-like syndrome (LLS) describes a mostly extravascular acquired hemolytic anemia and cytopenia, due to splenomegalic hypersplenism secondary to poikilocytosis, which partly resolved post-splenectomy.
The patient is a 32-year-old Caucasian female, previously described [4] to have LLS due to the variant m.10191T>C in MT-ND3. The mutation was detected in buccal mucosa cells. Heteroplasmy was not determined as it was not covered by insurance. The patient presented with anemia at the age of 25, in August 2015, when blood counts revealed a decreased hemoglobin (Hb) and hematocrit (HCT), with elevated erythrocyte sedimentation rate (ESR) (Table 1). However, complete blood count (CBC) from age 14 already showed Hb of 11 - 12 g/dL. In September 2015, the RBC, Hb, and HCT were low, and red cell distribution width standard deviation (RDW-SD) was elevated (Supplementary Material 1, www.thejh.org). Polyspecific direct Coombs antibody test was negative. Bone marrow biopsy (BMB) revealed 100% hypercellular marrow, erythroid hyperplasia and increased reticulin fibers. Despite the negative antibody test, the patient was placed on cyclosporine A (100 mg/day) for a presumptive autoimmune process.
 Click to view | Table 1. Relevant Parameters of the Anemia Workups |
In October 2015, haptoglobin was low. Paroxysmal nocturnal hemoglobinuria testing was negative and computed tomography (CT) revealed splenomegaly (Supplementary Material 1, www.thejh.org).
In January 2016, the RBC, HCT and Hb were lower compared to previous results. Coombs antibody was checked again and still negative, while lactic dehydrogenase was low for hemolysis. The patient received blood transfusion.
In February 2016, Hb, RBC, HCT, platelet (PLT) and WBC were all at their lowest points. Activated partial thromboplastin time was prolonged at 37 s (normal: 22 - 31 s). Another blood transfusion was given, a diagnosis of cytopenia secondary to hypersplenism was made, and the patient was taken off cyclosporine. ESR was extremely elevated. Schistocytes and other RBC morphology were negative. Glucose-6-phosphate dehydrogenase (G6PD) levels measured after transfusion were normal. Serum protein electrophoresis was normal except for low alpha-2. Immunoglobulins G, A and M, rheumatoid factor, antinuclear antibodies, and complements C3 and C4 were normal. BMB revealed 100% hypercellular marrow, erythroid hyperplasia, normal iron stores and no lymphoid aggregates or reticulin fibers.
The splenectomy was performed in May 2016. This choice was made because other diseases had been ruled out, along with poikilocytosis and the spleen’s continued growth with a trend toward pancytopenia. The size on CT was larger than normal gross pathological examination, which revealed an enlarged spleen weighing 404 g (normal female: 50 - 250 g), and spleen pathology revealed extramedullary hematopoiesis, congestion of the red pulp, scattered macrophages and follicular hyperplasia. The pathologist determined that the findings were consistent with acquired hemolytic anemia. The patient experienced severe thrombocytosis of 1,600 × 103/µL (normal: 150 - 450 × 103/µL), but examinations for thrombosis were negative (Supplementary Material 1, www.thejh.org).
In September 2016, haptoglobin was elevated, RBC, Hb and HCT had improved, WBC had fully recovered, and thrombocytosis had lowered. The RDW-SD had increased even higher than the peak of hemolysis in February 2016. Additionally, RBC morphology revealed 3+ anisocytosis, 3+ macrocytosis, +1 elliptocytosis and +1 schistocytosis (Fig. 1). A final BMB surprisingly still revealed a 100% hypercellular marrow. Reticulin was normal and iron storage was depleted. Lymphoid aggregates of polyclonal mixed T and B cells were detected. The myeloid to erythroid ratio increased from 1:2 to 1:1, indicating a reduction in erythroid precursors due to mitigation of secondary destruction. Peripheral slide examinations revealed mixed poikilocytes, including unreported “clover” cells which appear to be two RBC crossed over one another (Fig. 1). The whole exome sequencing (WES) ruled out hexokinase and pyruvate kinase deficiencies (Supplementary Material 1, www.thejh.org).
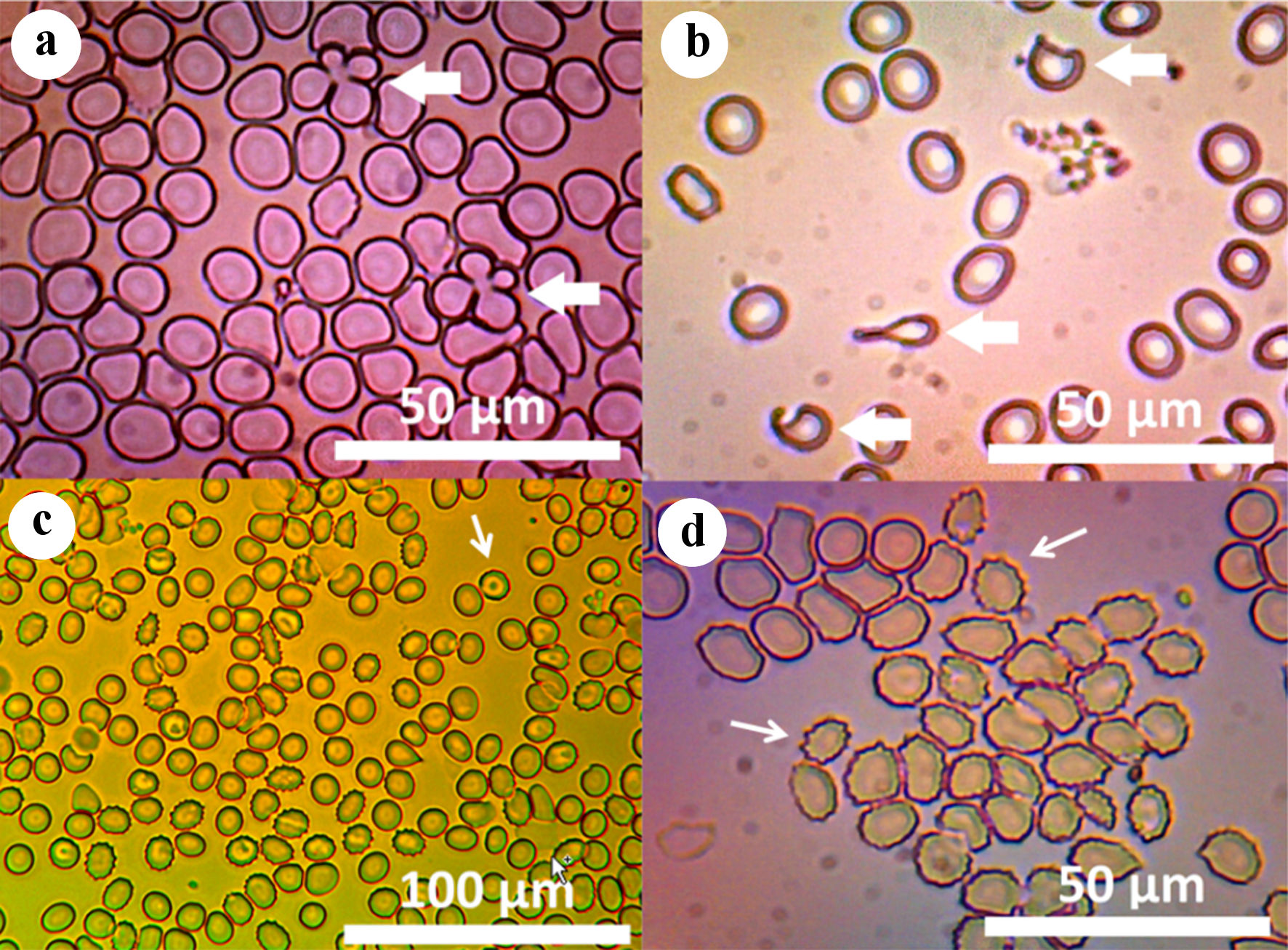 Click for large image | Figure 1. (a) Micrograph of unstained peripheral blood viewed with compound light microscopy through 606 - 660 nm red filter, demonstrating unreported “clover” cells, two RBCs folded over one another (× 1,000, arrows). (b) Micrograph of eosin Y-stained peripheral blood viewed with compound light microscopy demonstrating dacrocytes (× 1,000, arrows). (c) Micrograph of peripheral blood smear stained with acridine orange, viewed under fluorescence with 450 - 490 nm IVFL epi-fluorescence condenser, demonstrating echinocytosis and scattered “target cells” (× 400, arrows). (d) Micrograph of unstained peripheral blood viewed with compound light microscopy demonstrating echinocytosis (× 1,000, arrows). RBC: red blood cell. |
In October 2017, blood counts revealed normal RBC, but Hb and HCT were low, with mean corpuscular hemoglobin (MCH) now low. Additionally, RBC morphology on a 300-cell slide revealed 75% anisocytosis, 75% macrocytosis, 25% elliptocytosis and 25% schistocytosis (Fig. 1). High-performance liquid chromatography (HPLC) hemoglobinopathy panel was normal. The patient was 99.5% Western European.
In May 2018, WBC was elevated, and Hb and iron were low, with normal total iron-binding capacity (TIBC). The patient was started on ferrous gluconate. In October 2018, RBC had risen and Hb was low-normal, but MCH was still low. Iron was normal with elevated TIBC.
The patient’s highest Hb was 13.1 g/dL (normal: 12.1 - 15.1 g/dL) in April 2019, but in August 2021, this had lowered, PLT count was the lowest since splenectomy, and MCH was normal. In April 2023 RBC, Hb, and HCT were normal, but thrombocytosis persisted.
This case shows that LLS due to the variant m.10191T>C in MT-ND3 can manifest with extravascular hemolytic anemia due to poikilocytosis. Hemolytic anemia is a rare manifestation of MIDs, but other types of anemia are known phenotypic features of syndromic and non-syndromic MIDs [5-7], with sideropenic anemia being the most commonly reported.
The final diagnosis in the index patient, however, was hemolytic anemia due to nonspecific poikilocytosis and cytopenia secondary to hypersplenism. It is likely that the poikilocytes induced hypersplenism. Though not a typical case of hypersplenism, many features indicated a need for splenectomy, agreed upon by several hematologists. Notably, the blood counts kept dropping, precipitating symptoms of opportunistic infection, petechiae, easy bruising and anemia. The patient experienced modest improvement after splenectomy and has not had any blood transfusions since then. The patient also has recurrent lymphadenopathy, thymic hyperplasia, B-cell lymphocytosis, hypogammaglobulinemia, absence of plasma cells on BMB, germinal center hyperplasia, elevated inflammatory markers, elevated C3 and variants of unknown significance (VUS) in STAT4.
Every known cause of hemolytic anemia was excluded in the index patient, including paroxysmal nocturnal hemoglobinuria (PNH), autoimmune hemolytic anemia, cold antibody hemolytic anemia, G6PD deficiency, while WES ruled out hexokinase deficiency, pyruvate kinase deficiency, hereditary RBC membrane diseases, and hereditary poikilocytoses. WES was repeated, increasing its reliability. The causative variant was also detected in the index patient’s mother. Hemoglobinopathies, thalassemias, poisoning and drug side effects were also ruled out.
Of interest is the unusual number of types of poikilocytes, as well as the novelty of some of their shapes, some of which are not previously described. Also of interest are the low reticulocytes, lactate dehydrogenase (LDH) and bilirubin in a state of pseudo-hemolysis. Haptoglobin immediately resolved post-splenectomy. It is also unknown why the bone marrow remained 100% hypercellular after splenectomy, and why there were schistocytes present for years, unless there was an intravascular component, too. There also could be ineffective erythropoiesis, though the WBC count resolved immediately post-splenectomy. However, WES revealed no mutations for a myelodysplastic process, and none was detected via fluorescence in situ hybridisation (FISH) and karyotyping on BMB. Though myelodysplastic processes have a mitochondrial element [8], and low haptoglobin with splenomegaly can occur during myelodysplastic syndrome (MDS), there is no evidence of MDS, nor would it be expected to resolve any symptoms and blood counts after splenectomy.
The patient was diagnosed with LLS due to m.10191T>C in MT-ND3. Increased RBC destruction by splenic macrophages in mtDNA mutations has been previously described in mice and is thought to be a result of ineffective purging of mtDNA as RBCs mature, affecting their cell membrane shapes [9]. Pearson’s syndrome is an mtDNA disorder which manifests with anemia and increased heteroplasmy in leukocytes [10, 11], but the patient does not have the typical findings of Pearson’s syndrome including vacuolization of bone marrow precursors, nor reticulocytosis. It is currently unknown why some MID patients present with certain hematological manifestations and others do not, as well as most other symptoms. However, there are indications that the respiratory chain is essential for hematopoietic stem cell function [12]. There are also indications that mtDNA variants impair elimination of mitochondria during erythroid maturation, leading to enhanced erythrocyte destruction [13]. However, no unique morphology of erythroid precursors was noted on BMB.
Whether or not there is a relation between hematology and immunology remains speculative. The high ESR could be due to recurrent infections. High ESR and C-reactive protein (CRP) were always a feature during “critical illness” and have happened in the absence of the described hematological phenomena. Though, the ESR immediately went down post-splenectomy. There were recurrent infections due to cyclosporine usage.
In conclusion, this case is, to our knowledge, the first described case of hemolytic anemia due to poikilocytosis related to mtDNA disease. More study is warranted to understand any role of mtDNA in poikilocytoses in MIDs.
| Supplementary Material | ▴Top |
Suppl 1. Graphical depiction of the timeline and disease course.
Acknowledgments
None to declare.
Financial Disclosure
No funding was received.
Conflict of Interest
None to declare.
Informed Consent
Not applicable.
Author Contributions
SMN: design, literature search, discussion, first draft, critical comments, and final approval. JF: literature search, discussion, critical comments, second draft, and final approval.
Data Availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
| References | ▴Top |
- Baldwin C, Pandey J, Olarewaju O. Hemolytic Anemia. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023.
pubmed - Bandaru SS, Killeen RB, Gupta V. Poikilocytosis. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023.
pubmed - Arias CF, Arias CF. How do red blood cells know when to die? R Soc Open Sci. 2017;4(4):160850.
doi pubmed pmc - Newstead SM, Finsterer J. Leigh-like syndrome with a novel, complex phenotype due to m.10191T>C in Mt-ND3. Cureus. 2022;14(9):e28986.
doi pubmed pmc - Oncul U, Unal-Ince E, Kuloglu Z, Teber-Tiras S, Kaygusuz G, Eminoglu FT. A novel PUS1 mutation in 2 siblings with MLASA syndrome: a review of the literature. J Pediatr Hematol Oncol. 2021;43(4):e592-e595.
doi pubmed - Tesarova M, Vondrackova A, Stufkova H, Veprekova L, Stranecky V, Berankova K, Hansikova H, et al. Sideroblastic anemia associated with multisystem mitochondrial disorders. Pediatr Blood Cancer. 2019;66(4):e27591.
doi pubmed - Ying Y, Liang Y, Luo X, Wei M. Case report: clinical and genetic characteristics of pearson syndrome in a Chinese boy and 139 patients. Front Genet. 2022;13:802402.
doi pubmed pmc - Nasr W, Filippi MD. Acquired and hereditary bone marrow failure: A mitochondrial perspective. Front Oncol. 2022;12:1048746.
doi pubmed pmc - Ohkubo N, Suzuki Y, Aoto M, Yamanouchi J, Hirakawa S, Yasukawa M, Mitsuda N. Accelerated destruction of erythrocytes in Tie2 promoter-driven STAT3 conditional knockout mice. Life Sci. 2013;93(9-11):380-387.
doi pubmed - Son JS, Seo GH, Kim YM, Kim GH, Jin HK, Bae JS, Im HJ, et al. Clinical and genetic features of four patients with Pearson syndrome: An observational study. Medicine (Baltimore). 2022;101(5):e28793.
doi pubmed pmc - Uygun V, Daloglu H, Ozturkmen S, Karasu G, Yesilipek A. Pearson syndrome in a child transplanted for Diamond-Blackfan anemia. Arch Argent Pediatr. 2021;119(5):e559-e561.
doi pubmed - Anso E, Weinberg SE, Diebold LP, Thompson BJ, Malinge S, Schumacker PT, Liu X, et al. The mitochondrial respiratory chain is essential for haematopoietic stem cell function. Nat Cell Biol. 2017;19(6):614-625.
doi pubmed pmc - Ahlqvist KJ, Leoncini S, Pecorelli A, Wortmann SB, Ahola S, Forsstrom S, Guerranti R, et al. MtDNA mutagenesis impairs elimination of mitochondria during erythroid maturation leading to enhanced erythrocyte destruction. Nat Commun. 2015;6:6494.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.