

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://www.thejh.org |
Case Report
Volume 12, Number 5, October 2023, pages 227-230

A Rare Coexistence of Multiple Myeloma and Polycythemia Vera
Alexander Landsmana, c, Priyanka Baruab, Alida Podrumarb
aDepartment of Internal Medicine, Nassau University Medical Center, East Meadow, NY 11554, USA
bDepartment of Hematology & Oncology, Nassau University Medical Center, East Meadow, NY 11554, USA
cCorresponding Author: Alexander Landsman, Department of Internal Medicine, Nassau University Medical Center, East Meadow, NY 11554, USA
Manuscript submitted July 14, 2023, accepted September 4, 2023, published online October 21, 2023
Short title: Rare Coexistence of MM and PCV
doi: https://doi.org/10.14740/jh1167
| Abstract | ▴Top |
Multiple myeloma (MM) is classically associated with organ dysfunction leading to hypercalcemia, renal insufficiency, anemia and bone disease, known as the CRAB criteria. More than 70% of patients with MM present with anemia. Few rare case reports, however, have demonstrated the presentation of MM associated with polycythemia. We present an interesting case of a 65-year-old female who was initially diagnosed with monoclonal gammopathy of undetermined significance (MGUS) which progressed to smoldering myeloma and later developed into MM. The patient also had coexisting polycythemia vera (PCV). We discuss the typical patient presentations as well as the expanded diagnostic criteria for MM. The pathophysiology explaining the coexistence of polycythemia and MM will be explored as well.
Keywords: Multiple myeloma; Polycythemia vera; Anemia; Monoclonal gammopathy of undetermined significance; Smoldering myeloma
| Introduction | ▴Top |
Multiple myeloma (MM) is a neoplasm of plasma cell origin causing proliferation of monoclonal plasma cells in the bone marrow leading to production of abnormal proteins. This malignant plasma cell accumulation can lead to organ dysfunction including hypercalcemia, renal injury, anemia, and bone destruction (classically known as the CRAB criteria) [1]. MM accounts for approximately 2% of all diagnosed cancers in the United States and predominately occurs in adults aged 65 and older [2, 3]. The disease progresses as an insidious process typically with precursor conditions known as monoclonal gammopathy of undetermined significance (MGUS) or smoldering MM. These precursors do not have the signs or symptoms of organ dysfunction seen in MM or other lymphoproliferative diseases. Approximately 5% of patients with MGUS, and 50% of patients with smoldering myeloma will progress to develop MM within 5 years of diagnosis [1].
An analysis of over 1,000 MM patients between the years 1985 and 1998 showed anemia with hemoglobin levels less than 12 g/dL identified in 73% of the cohort [2]. Bone marrow invasion of the abnormal proteins, erythropoietin deficiency due to renal impairment, iron deficiency, and myelosuppression due to chemotherapy can all be factors contributing to the typical anemia seen as diagnostic of the MM disease course [4]. Several rare case reports, however, have demonstrated the uncommon concurrence of polycythemia with MM. There have been reports of patients having MM associated with both primary polycythemia vera (PCV) as well as secondary causes of polycythemia [5]. This case report highlights a patient who developed MM and was concurrently found to have primary PCV.
| Case Report | ▴Top |
Investigations
A 65-year-old female with a past medical history of hypertension, hyperlipidemia, endometrial carcinoma status post total abdominal hysterectomy with bilateral salpingo-oophorectomy (TAHBSO) and radiation therapy in 2009, thyroid follicular carcinoma status post thyroidectomy and radioactive iodine treatment in 2008 presented to the Hematology and Oncology clinic at Nassau University Medical Center (NUMC) to establish care in 2018 after moving to New York. The patient also reported that she had been diagnosed with MGUS in the past but never followed up for further testing. On presentation, the patient denied significant weight loss, night sweats, or any bone pain. She had a positive family history of leukemia in her father and sister. Physical examination on initial presentation was unremarkable.
Diagnosis
Initial blood and urine studies were ordered which revealed serum hemoglobin of 13.2 g/dL, calcium 8.4 mg/dL, creatinine 1.1 mg/dL, total protein 8.8 g/dL and albumin 3.3 g/dL. Both serum and urine protein electrophoresis revealed an M-spike with an abnormal protein band of 2.0 g/dL. Further analysis revealed elevated IgG levels at 2,550 mg/dL, low IgA (45.3 mg/dL) and IgM (5 mg/dL) levels. Serum immunofixation further identified an IgG lambda monoclonal protein. The kappa free light chain was 18.7 mg/L and lambda free light chain was 258 mg/L. The kappa/lambda free light chain ratio was 0.07, below the normal reference range, confirming the presence of monoclonal free light chains in the serum.
Bone marrow biopsy was performed in March of 2019 which revealed mild perivascular plasmacytosis with ranges from less than 5% to focally 10-15%. Flow cytometry showed clonal expansion of plasma cells with cytoplasmic lambda light chain restriction 0.03%. Fluorescence in situ hybridization (FISH) revealed a monosomy 13 and a gain of 1q on the chromosomal analysis. Based on the biopsy results, the patient was diagnosed with smoldering MM. Computed tomography (CT) imaging did not reveal any lytic bone lesions at that time.
Several months later in January of 2020, on routine blood testing the patient was noted to have hemoglobin of 17.9 g/dL and hematocrit of 57.4%. Further testing showed a low erythropoietin (EPO) level at 1.2 mIH/mL. JAK2 with V617F mutation was detected and the patient was diagnosed with primary PCV. According to the Molecular International Prognostic Scoring System for Polycythemia Vera (MIPSS-PV), the patient was found to have 1 point and stratified in the low-risk category. At the time of diagnosis, the patient also was found to have a pulmonary embolism (PE) on CT imaging.
Treatment
The patient was diagnosed with primary PCV and was treated with daily hydroxyurea and serial weekly phlebotomy sessions. Complete blood count (CBC) and other blood tests were monitored closely. Eliquis was additionally initiated as treatment for PE found on CT imaging.
Follow-up and outcomes
Later in the disease course, the patient’s blood work was noted to have an elevated M-spike on protein studies. Bone marrow biopsy was repeated in September of 2021, revealing total plasma cells comprising about 10-20% of the total marrow cells (Fig. 1). Magnetic resonance imaging (MRI) of the pelvis was performed in October of 2021, revealing diffuse heterogeneous marrow with a suspicious focal marrow replacement of 2.3 cm in the posterior iliac crest. Based on the MRI findings and the biopsy report, the patient was diagnosed with MM. Further testing showed a serum beta-2 microglobulin value of 5.45 mg/L and serum albumin of 3.8 g/dL, indicating an International Staging System (ISS) stage III. She was then started on treatment with daily lenalidomide as well as weekly dexamethasone. The patient is currently undergoing continued treatment for MM. Hemoglobin levels however have normalized and treatment for polycythemia has completed. Subsequently, after initiating care in our facility, the patient had relocated and decided to continue treatments with a private hematologist-oncologist in another state.
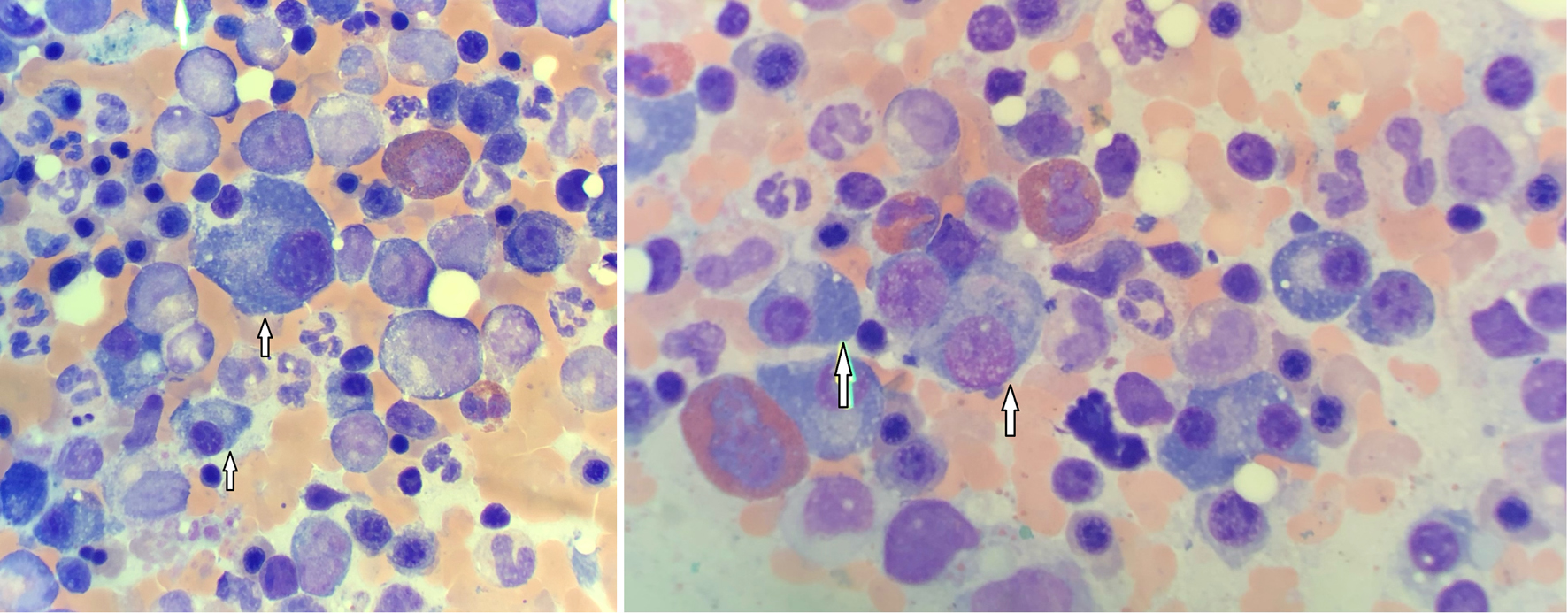 Click for large image | Figure 1. Bone marrow biopsy showing numerous plasma cells (arrows). |
| Discussion | ▴Top |
Patients with MM can initially present with laboratory abnormalities such as anemia, hypercalcemia, elevated creatinine, or proteinuria. Symptoms on evaluation may include nausea, vomiting, malaise, generalized fatigue, or weight loss. Patients may also present with bone pain due to underlying lytic lesions. After thorough history taking and physical examination is performed, patients with high clinical suspicion for MM should then undergo laboratory testing. Testing includes CBC with differential, evaluation of peripheral blood smear, complete chemistry panel, serum free light chain assay, serum protein electrophoresis (SPEP) with immunofixation, as well as urine collection for protein electrophoresis and immunofixation (UPEP) [6]. Further evaluation includes whole-body radiographic imaging as well as bone marrow biopsy.
In 2014, the International Myeloma Working Group (IMWG) expanded the diagnostic criteria required for MM. Bone marrow analysis must show greater than 10% abnormal plasma cells. Additionally, there must be evidence of a myeloma defining event as seen with either end organ damage or a myeloma biomarker. End organ damage can be one of the CRAB criteria as mentioned above: hypercalcemia, kidney injury, anemia, or an osteolytic lesion seen on imaging. The IMWG included the notion of a myeloma defining biomarker which can confirm the diagnosis in substitution for the CRAB criteria. The biomarkers include bone marrow analysis containing 60% or greater plasma cells, a ratio of involved to uninvolved free light chains being 100 or more, or at least one focal lesion seen on MRI that measures at least 5 mm in size [7].
More than 70% of MM patients present with characteristic anemia on laboratory testing. Several rare case reports, including ours, have demonstrated the possibility of MM with concurrent polycythemia. Polycythemia can be subdivided into primary and secondary causes. Primary polycythemia is caused by a mutation that leads to the proliferation of erythroid cells. PCV, the classical primary polycythemia, is usually caused by a somatic mutation (V617F) in the JAK2 gene. Secondary polycythemias, on the other hand, are caused by increased EPO production. Chronic hypoxia, areas with high altitudes or paraneoplastic syndromes are some of the many causes of increased EPO production that can be seen with secondary polycythemia.
One proposed explanation for the association of polycythemia and MM has been offered. The deposition of monoclonal light chains in the renal tubules causes damage to the renal cells decreasing oxygen delivery to the surrounding renal tissues. Transcription factors, known as hypoxia inducible factor (HIF) become activated causing upregulation of EPO which will increase production of red blood cells [5, 8]. This proposed mechanism however only provides insight for the association of MM with secondary polycythemia and elevated levels of EPO. Our patient, however, demonstrated diagnostic criteria for primary PCV with low EPO levels and JAK2 V617F mutation was identified. This theory is therefore less likely to explain the phenomenon in our patient.
A newly described, TEMPI syndrome must be considered when identifying a patient with coincidental erythrocytosis and a clonal plasma cell proliferative disorder. TEMPI syndrome includes telangiectasias, elevated EPO and erythrocytosis, monoclonal gammopathy, perinephric fluid collections, and intrapulmonary shunting. TEMPI syndrome has been described as a plasma cell neoplasm with associated paraneoplastic manifestations. The pathogenesis remains unclear, but it is hypothesized that the monoclonal protein triggers a paraneoplastic syndrome leading to increased EPO production. Of the few cases of TEMPI syndrome identified thus far, there have been extremely elevated EPO levels with negative JAK2 V617F mutations in all such cases. Additionally, only one such case with TEMPI syndrome has developed smoldering myeloma, the others with only MGUS [9]. Our patient had low EPO levels, mutation in the JAK2 gene, and displayed plasma cell expansion beyond MGUS, making the diagnosis of TEMPI less likely.
It has been suggested that both PCV and MM are clonal disorders with hematopoietic stem cell origin [10]. PCV is described as a myeloproliferative neoplasm with stem cell expansion of erythrocytes from the myeloid lineage. On the other hand, MM is known as a plasma cell dyscrasia with proliferation of plasma cells from the lymphoid lineage. A recent study analyzed two different patients both diagnosed with JAK2 mutation positive PCV and each later developed smoldering myeloma. Interestingly, the JAK2 V617F mutation was detected in the bone marrow plasma cells of these two patients as well, suggesting a single driver mutation for both the plasma cell and erythrocyte proliferations in these cases [11]. It is possible that patients with concurrent PCV and MM have a common pluripotent hematopoietic stem cell dysfunction causing proliferative diseases in both myeloid and lymphoid cell lines [10, 12]. This hypothesis is the most relevant and likely possible explanation for the disease concurrence in our patient. Dysfunction in our patient presented above may have originated from early pluripotent progenitor hematopoietic stem cells leading to both primary PCV and MM. Chromosomal and stem cell analysis would be required to confirm this hypothesis.
Myeloproliferative neoplasms (MPNs) as a whole have reportedly been associated with plasma cell dyscrasias including MGUS and MM [13]. MPN disorders are described as an excess production of hematopoietic stem cells in the bone marrow. These include PCV, essential thrombocythemia, and primary myelofibrosis. One retrospective study analyzed 90 patients with known MPN disorders. Out of these 90 patients, 32 of them had documented immunofixation results. Of these 32 patients, 15 were identified as having either MGUS or MM [14]. The increased association between MPN disorders and plasma cell dyscrasias may be explained by the above hypothesis as well. Hematopoietic stem cell dysfunction may give simultaneous rise to disease in both myeloid and lymphoid cell lines.
Our patient was found to have PCV with a lower-than-normal range level of EPO. The precise pathophysiology to explain the association between primary PCV and MM remains unclear. Further clinical research and analysis is required to provide a better explanation of this uncommon occurrence.
Learning points
MM is a plasma cell neoplasm with propensity for multiple organ system dysfunction. Routine blood tests with CBC and hemoglobin level should be monitored for development of anemia. Anemia will be a likely finding in those patients diagnosed with MM and should be addressed and managed appropriately. This case report, however, suggests the possibility of polycythemia as a simultaneous finding with MM. Awareness of this unlikely concurrence will allow for improved care with early diagnosis and prompt treatments.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
Informed patient consent was obtained.
Author Contributions
Alexander Landsman contributed to the literary search and wrote the main manuscript text. Priyanka Barua prepared figures and contributed to revisions and edits of the manuscript. Alida Podrumar contributed to revisions and offered the final approval of the manuscript. All authors reviewed the manuscript.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- van de Donk N, Pawlyn C, Yong KL. Multiple myeloma. Lancet. 2021;397(10272):410-427.
doi pubmed - Kyle RA, Gertz MA, Witzig TE, Lust JA, Lacy MQ, Dispenzieri A, Fonseca R, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78(1):21-33.
doi pubmed - Myeloma - Cancer Stat Facts. SEER. https://seer.cancer.gov/statfacts/html/mulmy.html.
- Cline MJ, Berlin NI. Studies of the anemia of multiple myeloma. Am J Med. 1962;33:510-525.
doi pubmed - Hutchison EJ, Taverna JA, Yu Q, Yeager AM. Polycythaemia: an unusual presentation of multiple myeloma. BMJ Case Rep. 2016;2016.
doi pubmed pmc - Cowan AJ, Green DJ, Kwok M, Lee S, Coffey DG, Holmberg LA, Tuazon S, et al. Diagnosis and management of multiple myeloma: a review. JAMA. 2022;327(5):464-477.
doi pubmed - Rajkumar SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G, Mateos MV, Kumar S, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538-548.
doi pubmed - Shu S, Wang Y, Zheng M, Liu Z, Cai J, Tang C, Dong Z. Hypoxia and hypoxia-inducible factors in kidney injury and repair. Cells. 2019;8(3):207.
doi pubmed pmc - Rosado FG, Oliveira JL, Sohani AR, Schroyens W, Sykes DB, Kenderian SS, Lacy MQ, et al. Bone marrow findings of the newly described TEMPI syndrome: when erythrocytosis and plasma cell dyscrasia coexist. Mod Pathol. 2015;28(3):367-372.
doi pubmed - Raskind WH, Jacobson R, Murphy S, Adamson JW, Fialkow PJ. Evidence for the involvement of B lymphoid cells in polycythemia vera and essential thrombocythemia. J Clin Invest. 1985;75(4):1388-1390.
doi pubmed pmc - Lee H, McCulloch S, Mahe E, Shafey M, Rashid-Kolvear F, Khan F, Prajapati D, et al. Anti-myeloma potential of ruxolitinib in co-existing JAK2V617F-positive smouldering myeloma and polycythaemia vera. Br J Haematol. 2020;189(3):e114-e118.
doi pubmed - Fink L, Bauer F, Perry JJ. Coincidental polycythemia vera and multiple myeloma: case report and review. Am J Hematol. 1993;44(3):196-200.
doi pubmed - Economopoulos T, Papageorgiou S, Pappa V, Papageorgiou E, Valsami S, Kalantzis D, Xiros N, et al. Monoclonal gammopathies in B-cell non-Hodgkin's lymphomas. Leuk Res. 2003;27(6):505-508.
doi pubmed - Malhotra J, Kremyanskaya M, Schorr E, Hoffman R, Mascarenhas J. Coexistence of myeloproliferative neoplasm and plasma-cell dyscrasia. Clin Lymphoma Myeloma Leuk. 2014;14(1):31-36.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.