

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://www.thejh.org |
Case Report
Volume 13, Number 3, June 2024, pages 116-120

A Unique Case of a Compound Heterozygosity of Hemoglobin Korle-Bu and Sickle Cell Trait in a Military Trainee
Gartrell C. Bowlinga, b, Niels A. Rydena, c, Allen R. Holmesc, Lauren E. Leec, Kristin Stolla, c, d
aSan Antonio United Services Health Education Consortium, Brooke Army Medical Center, Fort Sam Houston, TX 78234, USA
bSchool of Medicine, Uniformed Services University, Bethesda, MD 20814, USA
cDepartment of Medicine, Brooke Army Medical Center, Fort Sam Houston, TX 78243, USA
dCorresponding Author: Kristin Stoll, Division of Hematology Oncology, Department of Medicine, Brooke Army Medical Center, Fort Sam Houston, TX 78234, USA.
Manuscript submitted February 27, 2024, accepted April 8, 2024, published online June 28, 2024
Short title: Compound Hb KB Heterozygosity With Hb S
doi: https://doi.org/10.14740/jh1257
| Abstract | ▴Top |
Hemoglobin Korle-Bu (Hb KB) is a rare and likely under-reported hemoglobin (Hb) variant resulting from an unusual point mutation on the beta-globin chain. Hb KB is typically clinically silent, and there are limited reports of Hb KB heterozygosity compounded with other hemoglobinopathies that can present with varying clinical phenotypes. Here, we report a case of compound Hb KB heterozygosity with Hb S in an asymptomatic military trainee with a positive sickle cell screening test. Hb capillary and gel electrophoresis predicted a compound Hb S/D-Punjab overlap, which foretells a severe clinical phenotype. Sequencing of the Hb beta gene HBB demonstrated Hb KB, allowing for a diagnosis that fit his asymptomatic clinical phenotype and allowed for retention in the military.
Keywords: Hemoglobin Korle-Bu; Sickle cell trait; Hemoglobin subunit beta sequencing
| Introduction | ▴Top |
Hemoglobin (Hb) subunit beta (HBB) is a globin protein component of the Hb tetrameter coded by the HBB gene on chromosome 11. Variations of HBB gene at the transcriptional or translational level affect the stability and the production of the beta globin chain leading to a wide array of Hb variants that may manifest with symptomatic presentations or remain clinically silent. In fact, there are over 950 deletions, point mutations, and other variations in HBB recorded in the ClinVar and HbVar databases [1, 2].
The most prevalent and widely recognized HBB variant is Hb S, which occurs when valine is substituted for the normal glutamic acid at the seventh amino acid [3]. This HBB mutation is responsible for sickle cell disease (SCD), a spectrum of inherited disorders associated with abnormal or “sickled” erythrocytes under conditions of deoxygenation [4]. Individuals who inherit a normal HBB allele and one abnormal allele encoding Hb S are known to carry the sickle cell trait (Hb AS; SCT), which is most often clinically silent [4]. However, those individuals homozygous for Hb S are known to have sickle cell anemia (Hb SS), which presents clinically with sickling events leading to hemolytic anemia, end-organ damage including pulmonary hypertension and retinopathy, splenic infarct, vaso-occlusive pain, and shortened overall survival [3, 4]. There are other compound heterozygous SCD genotypes involving Hb S with other inherited variants, such as Hb C, Hb D, Hb E, Hb H, and in this case report, Hb Korle-Bu (Hb KB) [4-7].
Hb KB is a rare beta globin variant that results from a HBB mutation at position 74, yielding a single amino acid substitution of aspartic acid with asparagine (HBB NM_000518.4:c.220G>A (p.Asp74Asn)) [5-8]. On Hb electrophoresis, Hb KB can be mistaken for several other beta chain hemoglobinopathies, including the most common Hb D variants: Hb D-Punjab (also known as Hb D-Los Angeles) and Hb D-Ibadan [9-11]. Cases of Hb S/D-Punjab have been associated with severe clinical phenotypes like that of Hb SS [12]. Conversely, in heterozygosity cases of Hb KB with other hemoglobinopathies, the phenotype may vary on the spectrum of asymptomatic to symptomatic [5, 6, 8, 12-15]. Unfortunately, there is paucity of literature available regarding Hb KB and its overlap syndrome, so understanding clinical manifestations are limited to case reports and small case series [6, 12, 16, 17]. Here, we illustrate the case of an active-duty service member with a clinically silent compound heterozygosity of Hb S and Hb KB variant who presented following a positive sickle cell screening test, now required of all incoming trainees.
| Case Report | ▴Top |
Investigations
A 19-year-old active-duty trainee with no significant past medical history was referred to Hematology following a positive sickle cell screen. He presented to the clinic in his usual state of health without acute complaint. He reported a normal, healthy childhood without recurrent illnesses and was physically active without limitations participating in both track and soccer. He denied a history of anemia, blood transfusions, and prior evaluation by a hematologist. His family heritage was of West African descent. He denied a family history of bleeding and clotting disorders, however, his father reported having been told he had sickle cell trait.
Diagnosis
The patient’s vitals were within normal limits, including a normal oxygenation saturation of 99% on room air. His physical exam was unremarkable and consistent with a young, healthy trainee. His complete blood count revealed a normal Hb of 15.3 g/dL and a mild microcytosis with mean corpuscular volume of 79 fL. His white blood cell count was normal at 4.2 × 103/µL with a normal differential and a normal platelet count of 235 × 103/µL. His complete metabolic panel was unremarkable, as were a lactate dehydrogenase (LDH) of 162 U/L and a haptoglobin of 32 g/dL. His peripheral blood smear (Fig. 1) was notable for erythrocytes with normal appearing morphology without target or sickled cells.
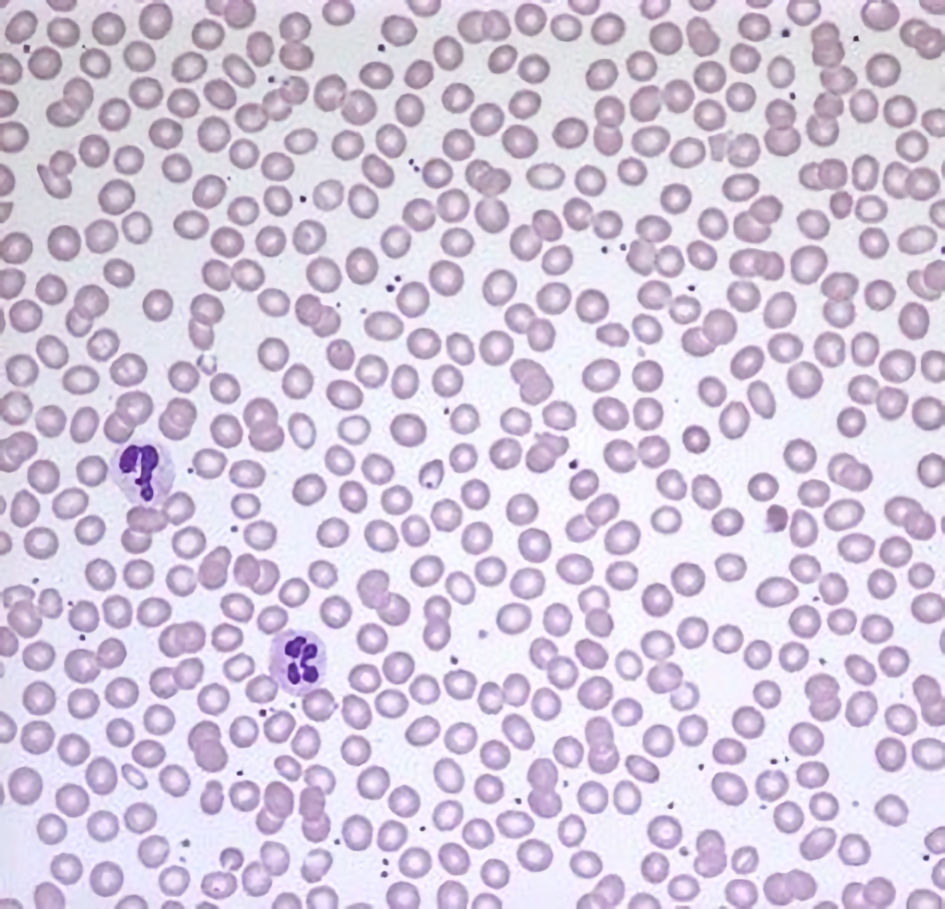 Click for large image | Figure 1. A peripheral blood smear with normochromic, normocytic erythrocytes without morphologic abnormality. |
Hb capillary electrophoresis was performed and demonstrated Hb A 0.0% (normal 95-98%); Hb F 0.6% (normal 0-2%); Hb S 41.4% (normal 0%), and Hb A2 5.1% (normal 0-2%). The sample was reflexed to Hb solubility and high-pressure liquid chromatography (HPLC) method for confirmation. HPLC demonstrated a Hb variant of 52.9% (normal 0%). The hematopathologist interpretation reads as abnormal with Hb pattern and concentration consistent with Hb D variant. His Hb S at 41% with a 53% Hb D variant would represent SCD with a compound heterozygous hemoglobinopathy of Hb S-Hb D. Given the pronounced lack of historical and laboratory evidence to support SCD and a grossly normal peripheral blood smear, Hb capillary electrophoresis was repeated. The results were similar with Hb A 0%; Hb S 43.4%; Hb A2 5.2%, and Hb other/variant of 51.4% (Fig. 2). The variant was again interpreted as Hb D or D-like peak based on the Hb gel electrophoresis agar (Fig. 3).
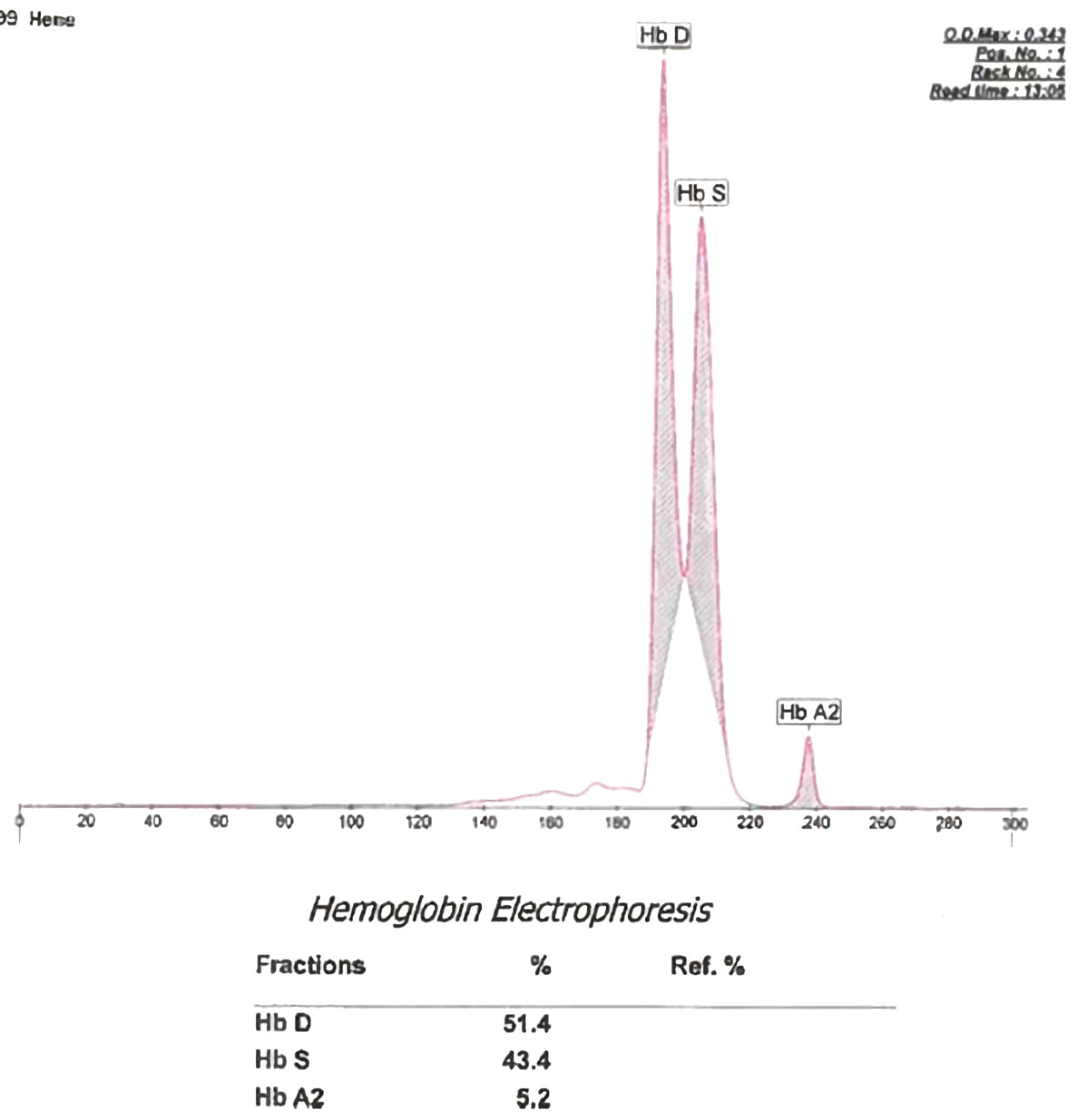 Click for large image | Figure 2. A hemoglobin (Hb) capillary electrophoresis showing Hb D variant, Hb S and Hb A2 peaks. |
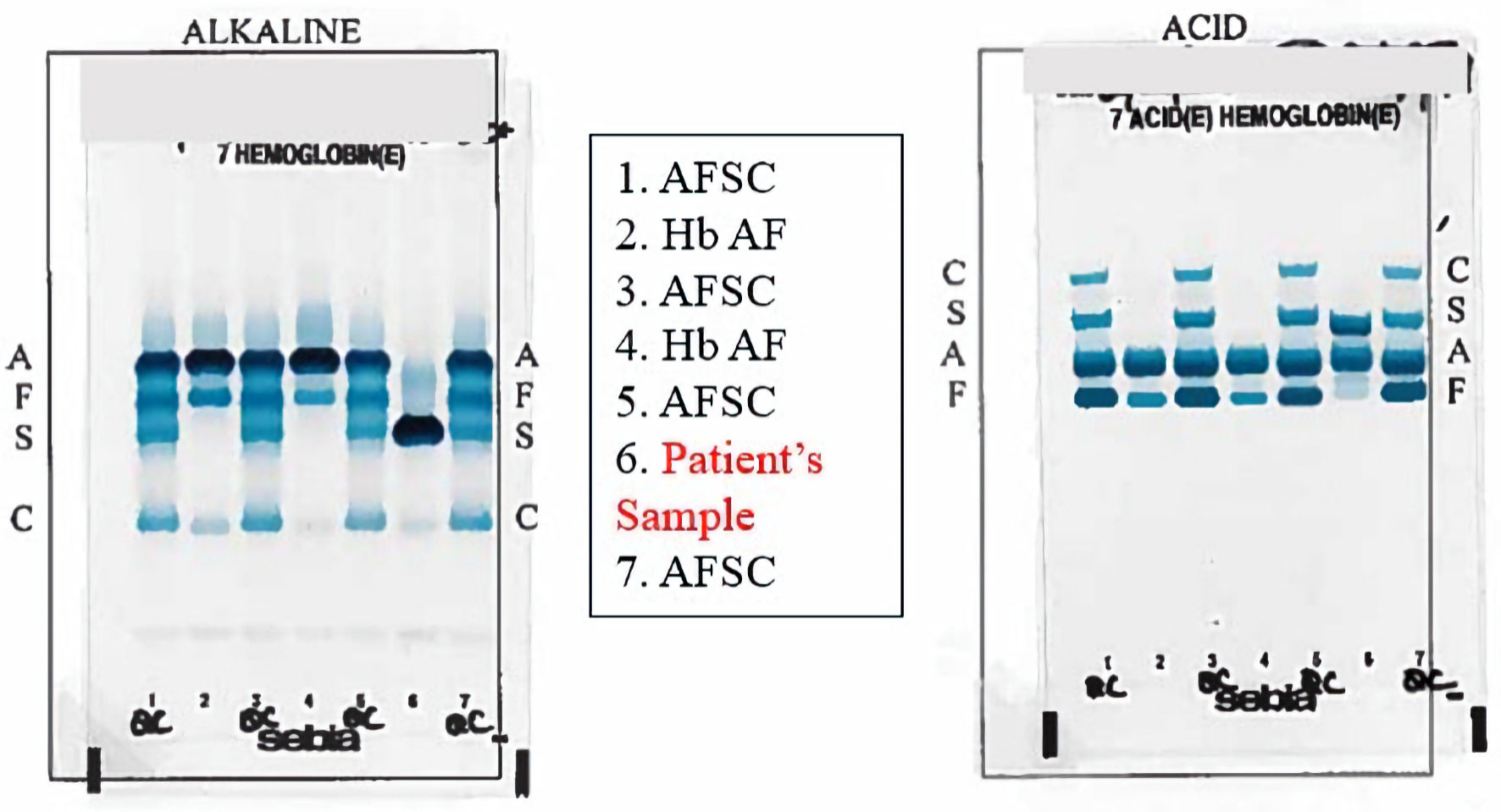 Click for large image | Figure 3. Acid and alkaline hemoglobin (Hb) gel electrophoresis showing a migration most consistent with Hb S, D, G, and Lepore in column 6 in both gels. AFSC controls in both gels are observed in columns 1, 3, 5, and 7. Hb AF comparisons in both gels are observed in columns 2 and 4. |
Carrying a diagnosis of a hereditary anemia such as SCD does not meet the accession standards outlined in the Department of Defense Instructions 6130.03, Volume 2 Medical Standards for Military Service: Retention [18]. Accordingly, evidence of hereditary anemia would disqualify the trainee from service with career implications for his future. Given the incongruence between the patient presentation and the severe phenotype that characterizes Hb S-Hb D compound heterozygosity, next-generation HBB genetic sequencing with deletion and duplication analysis was obtained via Fulgent Genetics (Fulgent Genetics, Inc.). Sequencing results did not reveal any known clinically significant variants; however, the test did identify two variants with abnormal point mutations associated with potential clinical relevance. The first HBB allele had a glutamic acid substituted for valine at codon 7 in exon 1 (HBB NM_000518.5:c.20A>T (p.Glu7Val)), which is consistent with Hb S. The second allele had an aspartic acid substituted for asparagine at codon 74 of exon 2 (HBB NM_000518.5:c.220G>A (p.Asp74Asn)). This is a missense variant of unknown significance and has been described as Hb KB. Per the HBB genetic sequencing report, this variant had been reported in the heterozygous state in two unrelated individuals with a phenotype like sickle cell trait [6, 16].
Follow-up and outcomes
Genetic sequencing of the HBB gene allowed for a diagnosis of compound heterozygosity of Hb S-Hb KB. This diagnosis more consistently fits with the patient’s clinical phenotype of a healthy patient without evidence of hereditary anemia. Accordingly, a recommendation was made to retain the service member. The recommendation was upheld, and he was retained in the military. Follow-up labs continue to be without evidence of anemia, hemolysis, asplenia, sickling crisis or rhabdomyolysis for more than a year from diagnosis.
| Discussion | ▴Top |
The prevalence of Hb KB is unclear. According to a recent query of the Genome Aggregation Database (GnomAD), this allele variant has been observed at a frequency of less than 0.01% (7/140204) [19]. While most reports have commonly pinpointed West Africa as a likely origin, variants have also been reported in descendants of Central America, Spain, and Thailand [11-13, 17, 20, 21]. When one beta allele harbors the Hb KB variant and the second beta allele harbors the Hb S variant, it produces compound heterozygosity. This compound variant is infrequently recorded in the medical literature, and our understanding is limited to a few case reports and case series (the largest out of Brazil with eight patients with the Hb KB variant [12]). A population-based retrospective case series was undertaken screening 3,590,315 children for Hb S/D-Punjab [12]. In total, 21 children initially screened positive, but with HBB gene sequencing, eight were identified to have Hb KB, and 11 were identified to have Hb S/D-Punjab with two exclusions. Those patients with Hb KB-Hb S were asymptomatic and maintained Hb > 10 g/dL and normal oxygenation saturation averaging 97%. Further, they did not require transfusions, nor did they experience any episodes of sickling, pain crises, or asplenia. By comparison, the patients with Hb S/D-Punjab had Hb < 10 g/dL with an average of 8 g/dL, required transfusions and support for sickling crises, demonstrated asplenia, and had an average oxygen saturation at 91% consistent with a severe phenotype. The study represented the largest population data available and demonstrated the clinical silence of Hb KB-Hb S compound heterozygosity was comparable to that of SCT.
Hb KB-Hb S is a rare mimic of Hb S/D-Punjab on Hb capillary and gel electrophoresis because it runs within the same window as Hb D. Other than clinical phenotype, discerning Hb KB-Hb S from Hb S-Hb D overlap requires the use of HBB genotyping. In the case of this active-duty service member, correctly identifying the variant compound hemoglobinopathy allowed for retention based current accession standards. This case supports the use of genotyping patients with unusual Hb electrophoresis results in the absence of other clinical findings for a clinically significant hemoglobinopathy. This is particularly important because as the military continues to perform sickle cell screening on each recruit, it is highly likely there will be an increased number of service members with these clinically silent HBB variations.
Learning points
This case report describes a novel variant of heterozygous sickle cell trait and Hb KB in an asymptomatic 19-year-old male. This diagnosis requires the use of HBB sequencing. As such, this case merits the use of Hb genotyping in patients with an unusual hemoglobinopathy pattern.
Acknowledgments
The opinions and assertions expressed herein are those of the author(s) and do not necessarily reflect the official policy or position of the Uniformed Services University of the Health Sciences or the Department of Defense.
Financial Disclosure
None to declare.
Conflict of Interest
Authors have no conflict of interest to disclose.
Informed Consent
Informed consent for publication from the patient was obtained.
Author Contributions
All authors contributed to the literature review, writing of original draft, and editing of original draft. KS was responsible for conceptualization and supervision.
Data Availability
Data and materials are available from the corresponding author on reasonable request.
| References | ▴Top |
- Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, Maglott DR. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42(Database issue):D980-985.
doi pubmed pmc - Hardison RC, Chui DH, Giardine B, Riemer C, Patrinos GP, Anagnou N, Miller W, et al. HbVar: A relational database of human hemoglobin variants and thalassemia mutations at the globin gene server. Hum Mutat. 2002;19(3):225-233.
doi pubmed - Arishi WA, Alhadrami HA, Zourob M. Techniques for the detection of sickle cell disease: a review. Micromachines (Basel). 2021;12(5):519.
doi pubmed pmc - Piel FB, Steinberg MH, Rees DC. Sickle cell disease. N Engl J Med. 2017;376(16):1561-1573.
doi pubmed - Adachi K, Kim J, Kinney TR, Asakura T. Effect of the beta 73 amino acid on the hydrophobicity, solubility, and the kinetics of polymerization of deoxyhemoglobin S. J Biol Chem. 1987;262(22):10470-10474.
pubmed - Nagel RL, Lin MJ, Witkowska HE, Fabry ME, Bestak M, Hirsch RE. Compound heterozygosity for hemoglobin C and Korle-Bu: moderate microcytic hemolytic anemia and acceleration of crystal formation [corrected]. Blood. 1993;82(6):1907-1912.
pubmed - Nagel RL, Johnson J, Bookchin RM, Garel MC, Rosa J, Schiliro G, Wajcman H, et al. Beta-chain contact sites in the haemoglobin S polymer. Nature. 1980;283(5750):832-834.
doi pubmed - Dolezal D, et al. Hemoglobin C/Korle-Bu identified in a patient with a previous diagnosis of hemoglobin S/C disease. American Journal of Clinical Pathology. 2020;154(Supplement_1):S110-S111.
- Torres Lde S, Okumura JV, Silva DG, Bonini-Domingos CR. Hemoglobin D-Punjab: origin, distribution and laboratory diagnosis. Rev Bras Hematol Hemoter. 2015;37(2):120-126.
doi pubmed pmc - Watson-Williams EJ, Beale D, Irvine D, Lehmann H. A New Haemoglobin, D Ibadan (Beta-87 Threonine — Lysine), Producing No Sickle-Cell Haemoglobin D Disease with Haemoglobin S. Nature. 1965;205:1273-1276.
doi pubmed - Konotey-Ahulu FI, Gallo E, Lehmann H, Ringelhann B. Haemoglobin Korle-Bu (beta 73 aspartic acid replaced by asparagine) showing one of the two amino acid substitutions of haemoglobin C Harlem. J Med Genet. 1968;5(2):107-111.
doi pubmed pmc - Rezende Pdo V, Costa Kda S, Domingues Junior JC, Silveira PB, Belisario AR, Silva CM, Viana MB. Clinical, hematological and genetic data of a cohort of children with hemoglobin SD. Rev Bras Hematol Hemoter. 2016;38(3):240-246.
doi pubmed pmc - Ropero P, Villegas A, Gonzalez FA. [Hemoglobin Korle-Bu [beta73(E17)Asp ->Asn]. First cases described in Spain]. Med Clin (Barc). 2004;123(7):260-261.
doi pubmed - Vukelja SJ. Hemoglobin Korle-Bu (G-ACCRA) in combination with hemoglobin C. Am J Hematol. 1993;42(4):412.
doi pubmed - Husan A, Amarasinghe SN, Fontenot A, Khan MW. Rare combinational hemoglobinopathies. Cureus. 2022;14(12):e32327.
doi pubmed pmc - Akl PS, Kutlar F, Patel N, Salisbury CL, Lane P, Young AN. Compound heterozygosity for hemoglobin S [beta6(A3)Glu6Val] and hemoglobin Korle-Bu [beta73(E17)Asp73Asn]. Lab Hematol. 2009;15(3):20-24.
doi pubmed - Honig GR, Seeler RA, Shamsuddin M, Vida LN, Mompoint M, Valcourt E. Hemoglobin Korle Bu in a Mexican family. Hemoglobin. 1983;7(2):185-189.
doi pubmed - DOD Instructions 6130.03, volume 2. Medical standards for military service: retention.
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, Collins RL, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434-443.
doi pubmed pmc - Siriratmanawong N, Chansri W, Singsanan S, Fucharoen G, Fucharoen S. Complex interaction of Hb E [beta26(B8)Glu—>Lys], Hb Korle-Bu [beta73(E17)Asp—>Asn] and a deletional alpha-thalassemia-1 in pregnancy. Hemoglobin. 2009;33(6):507-514.
doi pubmed - Changtrakun Y, Fucharoen S, Ayukarn K, Siriratmanawong N, Fucharoen G, Sanchaisuriya K. Compound heterozygosity for Hb Korle-Bu (beta(73); Asp-Asn) and Hb E (beta(26); Glu-Lys) with a 3.7-kb deletional alpha-thalassemia in Thai patients. Ann Hematol. 2002;81(7):389-393.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.