

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://www.thejh.org |
Case Report
Volume 13, Number 3, June 2024, pages 104-107

Fat Embolism Syndrome Mimicking Thrombotic Thrombocytopenic Purpura in a Patient With Hemoglobin S/Beta-Thalassemia
Bobby Sea, c, Austin Frischa, Min Woo Hwanga, Faran Polanib, Najeebah Badeb
aInova Fairfax Hospital Department of Internal Medicine, Falls Church, VA 22042, USA
bInova Schar Cancer Institute, Fairfax, VA 22031, USA
cCorresponding Author: Bobby Se, Inova Fairfax Hospital Department of Internal Medicine, Falls Church, VA 22042, USA
Manuscript submitted April 12, 2024, accepted May 28, 2024, published online June 28, 2024
Short title: FES Mimicking TTP
doi: https://doi.org/10.14740/jh1274
| Abstract | ▴Top |
Thrombotic microangiopathies cause ischemic organ damage and require urgent management for a favorable prognosis. Fat embolism syndrome from bone marrow necrosis is a rare and unique pathology that carries a high mortality rate. It can mimic thrombotic microangiopathies such as thrombotic thrombocytopenic purpura (TTP). Herein, we present a patient with sickle cell-beta-thalassemia who initially presented with a vaso-occlusive crisis, lab evidence of hemolysis, schistocytes and thrombocytopenia who developed acute encephalopathy with respiratory distress, consistent with TTP. She was found to have multiple infarcts in the brain. She was intubated and underwent plasma and red cell exchange. Bone marrow biopsy confirmed marrow necrosis from her vaso-occlusive crisis and subsequently, fat embolism syndrome. Here, we discuss the complex presentation and the complications of fat embolism from bone marrow necrosis and how it can mimic TTP.
Keywords: Fat embolism; Hemolytic anemia; Thalassemia; Thrombocytopenia
| Introduction | ▴Top |
Thrombotic microangiopathies (TMAs) are a group of disorders defined by the presence of microangiopathic hemolytic anemia, thrombocytopenia, and microthrombi, which lead to organ damage through ischemia. Thrombotic thrombocytopenic purpura (TTP) is a TMA pathology that requires quick identification and treatment to avoid severe complications and death [1]. TTP is defined by the presence of the classic TMA features of hemolytic anemia, schistocytes on peripheral smear, and thrombocytopenia in addition to low ADAMTS-13, and can present with signs of organ damage such as neurological impairment, renal dysfunction, or cardiac ischemia [2]. Similarly, fat embolism syndrome (FES) is characterized by pulmonary insufficiency, neurologic symptoms, thrombocytopenia, and anemia [3]. FES is most commonly seen after traumatic injuries such as long bone fractures, but other risk factors can include non-traumatic etiologies such as pancreatitis, liver disease, and sickle cell disease [4]. There have been case reports of TTP clinically mimicking FES and thus, making the diagnosis challenging [5]. Table 1 outlines the similarities and differences in the clinical and laboratory features between TTP and FES. We review the clinical presentation, pathophysiology, and current literature on the intersection between TTP and FES in sickle cell patients while highlighting the diagnosis and treatment needed to quickly differentiate these diseases and prevent death.
 Click to view | Table 1. Clinical and Laboratory Features Differentiating Between TTP and FES |
| Case Report | ▴Top |
Investigations
A 40-year-old female with a history of sickle-beta-thalassemia who presented to an outside hospital with severe back and bilateral lower extremity pain was transferred for management of acute sickle cell crisis. Upon arrival, lab work was largely unremarkable with hemoglobin of 12.8 g/dL, white blood cell counts of 12.9 × 103/µL, and platelet count of 253 × 103/µL. The patient was initially managed with fluids and pain medication for vaso-occlusive crisis but then developed acute encephalopathy. Follow-up lab work showed worsening hemolytic anemia and new onset thrombocytopenia with platelets of 105 × 103/µL. Laboratory testing showed elevated lactate dehydrogenase (LDH) to 1,070 U/L, low haptoglobin of < 8 mg/dL, elevated indirect bilirubin of 2.8 mg/dL and schistocytes were seen on the peripheral smear. She was intubated for airway protection given her worsening mental status and transferred to the intensive care unit (ICU). The following day, thrombocytopenia worsened to 79 × 103/µL. At this time, a computed tomography (CT) angiogram and CT of head were both negative. Her PLASMIC score was 6, which corresponds to a 72% risk of severe ADAMTS-13 deficiency. Therefore, the leading diagnosis at this time was TTP with other differential being fat embolism progressing to FES from non-traumatic bone marrow necrosis given the hemoglobinopathy and vaso-occlusive crisis.
Diagnosis
The patient underwent emergent plasma exchange and was started on high-dose steroids (1 g of solumedrol). Subsequently, ADAMTS-13 level was normal, and follow-up brain magnetic resonance imaging (MRI) showed multiple, scattered infarcts, highly suspicious for fat emboli. Bone marrow biopsy showed marrow necrosis (Fig. 1). Given these findings, FES became the established diagnosis.
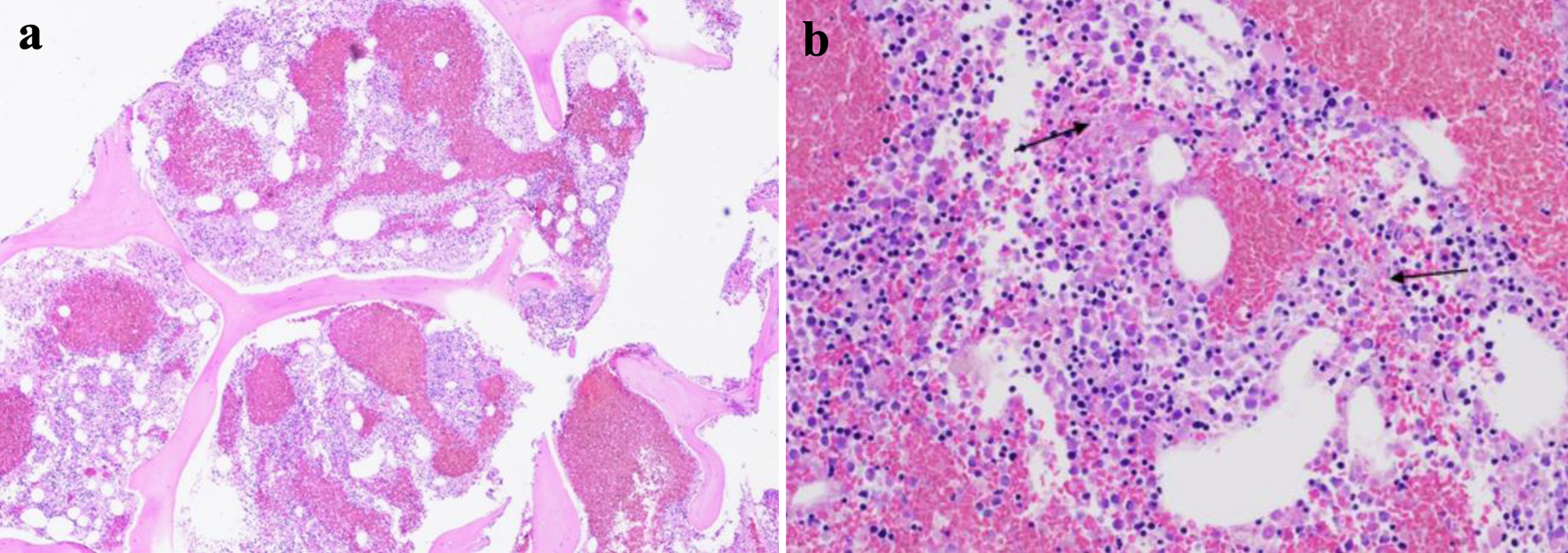 Click for large image | Figure 1. Bone marrow biopsy with evidence of bone marrow necrosis (a, × 4 magnification; b, × 20 magnification) (arrows). |
Treatment and outcomes
The patient therefore underwent red cell exchange (RCE) immediately which reduced the hemoglobin S levels to 17%. Throughout her stay in the ICU, the patient continued to have minimal neurologic recovery and electroencephalogram continued to show cerebral dysfunction in a non-specific manner. Hospital course was further complicated by continued fevers and tachycardia with leukocytosis prompting infectious workup and anti-microbial coverage. Her neurological status continued to show minimal recovery and eventually, tracheostomy and peg procedure were pursued prior to discharge to a long-term care facility.
Thrombocytopenia is defined as platelet levels < 150 × 103 U/L. Acute kidney injury is defined as an increase in serum creatinine by 0.3 mg/dL within 48 h, increase in serum creatinine > 1.5 times the baseline level within 7 days, or a decrease in urine output < 0.5 mL/kg/h for > 6 h. These definitions are consistent with the 2012 KDIGO clinical practice guidelines. Respiratory failure is defined as either hypoxemic or hypercapnic with PaO2 < 60 mm Hg or PaCO2 > 50 mm Hg, respectively. Altered mental status is defined as an acute change in cognitive function, psychological function, and/or level of consciousness in the form of behavior changes, alertness or confusion.
| Discussion | ▴Top |
The pathophysiology behind marrow necrosis causing FES is not yet fully understood. Classically, FES presents after long bone or pelvic trauma and only 10% of these cases have clinical manifestations such as anemia, thrombocytopenia, neurologic symptoms, and pulmonary insufficiency [6]. In patients with acute chest syndrome, circulating phospholipids from bone marrow necrosis have been found to activate the inflammatory cascade leading to fat emboli formation [7]. This observation agrees with the biochemical theory that explains non-traumatic causes of FES [8]. Therefore, patients with hemoglobinopathies are at increased risk for bone marrow necrosis leading to fat embolism and subsequently, FES [9]. Furthermore, there seems to be an increase in prevalence with sickle cell patients that have heterozygous genotypes compared to their homozygous counterparts. This correlation is poorly understood but patients with hemoglobin Sβ+ compared to SC and SS have a higher rate of developing FES [5, 9, 10]. The mortality rate for FES in the reported literature for sickle patients is 46%. Twenty percent of these patients that died also tested positive for parvovirus B19 [11, 12]. Therefore, hemoglobin Sβ+, SC and SS along with parvovirus are risk factors for the development of FES in patients with sickle cell disease.
While this disease is severe and life-threatening, diagnosing FES can be a challenge given clinically similar presentation as TTP. Distinguishing between the two diagnoses can be more complex in the setting of hemoglobinopathy due to chronic hemolysis. FES can rarely cause a triad of neurological impairment, thrombocytopenia, and organ failure making presentation nearly identical to TTP [13, 14]. Respiratory distress appears to be a distinguishing feature between the two as it is more common in FES; however, as demonstrated with our patient, relying on clinical presentation can be deceiving in these cases.
FES treatment is generally supportive care with a level of definitive management in sickle cell patients. This is because FES can cause acute chest syndrome in sickle cell patients, thus lending to RCE as an option. This exchange prevents the removed sickled cells from propagating more vaso-occlusive events and thus minimizing bone marrow necrosis [15]. Other treatment options include trialing high-dose steroids in patients with life-threatening FES which theoretically prevents more formation [16]. However, high-dose steroids can have adverse effects like infection like in our case thus making it a controversial treatment decision [17].
Given the high mortality rate of TTP and clinical findings in this case, it is reasonable to treat as TTP with plasma exchange while waiting on ADAMTS-13 levels on initial presentation. In the rare cases where FES was misdiagnosed as TTP, initial plasma exchange did not seem to negatively affect these patients [11, 18, 19]. However, it is important to re-evaluate the diagnosis based on clinical progression and new lab results. Here, our patient had a normal ADAMTS-13 level with MRI findings of multiple infarcts suggestive of fat embolization. Bone marrow biopsy to corroborate bone marrow necrosis confirmed our diagnosis of FES. While RCE is definitive in FES caused by sickle cell disease, the primary treatment modalities are supportive which further emphasizes the importance of prevention.
Learning points
FES resulting from bone marrow necrosis is an uncommon and distinctive pathology characterized by a significant mortality risk. Patients with sickle cell variant Hb Sβ+ who test positive for parvovirus B19 are at an increased risk of developing FES from bone marrow necrosis.
FES can imitate TMAs like TTP. Thus, plasma exchange is a reasonable initial therapy while waiting for ADAMTS-13 level to result before starting RCE.
Acknowledgments
None to declare.
Financial Disclosure
The authors have no funding source to disclose for this case report.
Conflict of Interest
The authors have no conflict of interest.
Informed Consent
Not applicable.
Author Contributions
Bobby Se: original draft preparation with literature review (lead); Austin Frisch: original draft preparation with literature review (support); Min Woo Hwang: review and editing; Faran Polani: review and editing; Najeebah Bade: review, editing, visualization of concept.
Data Availability
The authors declare that the data supporting the findings of this study are available withing the article.
| References | ▴Top |
- Arnold DM, Patriquin CJ, Nazy I. Thrombotic microangiopathies: a general approach to diagnosis and management. CMAJ. 2017;189(4):E153-E159.
doi pubmed pmc - Stanley M, Killeen RB, Michalski JM. Thrombotic thrombocytopenic purpura. In: StatPearls. Treasure Island (FL). 2024.
pubmed - Maitre S. Causes, clinical manifestations, and treatment of fat embolism. Virtual Mentor. 2006;8(9):590-592.
doi pubmed - Luff D, Hewson DW. Fat embolism syndrome. BJA Educ. 2021;21(9):322-328.
doi pubmed pmc - Ataga KI, Orringer EP. Bone marrow necrosis in sickle cell disease: a description of three cases and a review of the literature. Am J Med Sci. 2000;320(5):342-347.
doi pubmed - Kumar V, Fausto N, Abbas A. Robbins and Cotran’s pathologic basis of disease. 7th ed. New York, NY: WB Saunders. 2005; p. 137.
- Styles LA, Schalkwijk CG, Aarsman AJ, Vichinsky EP, Lubin BH, Kuypers FA. Phospholipase A2 levels in acute chest syndrome of sickle cell disease. Blood. 1996;87(6):2573-2578.
pubmed - Lehman EP, Moore RM. Fat embolism: including experimental production without trauma. Arch Surg. 1927;14(3):621-662.
- Tsitsikas DA, Gallinella G, Patel S, Seligman H, Greaves P, Amos RJ. Bone marrow necrosis and fat embolism syndrome in sickle cell disease: increased susceptibility of patients with non-SS genotypes and a possible association with human parvovirus B19 infection. Blood Rev. 2014;28(1):23-30.
doi pubmed - Saraf SL, Molokie RE, Nouraie M, Sable CA, Luchtman-Jones L, Ensing GJ, Campbell AD, et al. Differences in the clinical and genotypic presentation of sickle cell disease around the world. Paediatr Respir Rev. 2014;15(1):4-12.
doi pubmed pmc - Samaee S, Samaee S, Mihalca D, Fitzgerald L, Ahmed A, Hall J, Tsitsikas DA. Mortality Rates and autopsy findings in fat embolism syndrome complicating sickle cell disease. J Clin Pathol. 2023;76(7):497-500.
doi pubmed - Tsitsikas DA, Vize J, Abukar J. Fat embolism syndrome in sickle cell disease. J Clin Med. 2020;9(11):3601.
doi pubmed pmc - Tsitsikas DA, Mihalca D, Hall J, May JE, Gangaraju R, Marques MB, Scully M. Pitfalls in diagnosing thrombotic thrombocytopenic purpura in sickle cell disease. J Clin Med. 2022;11(22):235.
doi pubmed pmc - Rizvi S, Khakwani M, Pancham S, Tsitsikas D, Rudzki Z, Hassan-Smith G, Bowen M, et al. Bone marrow necrosis and fat embolism syndrome in sickle cell disease during COVID-19 infection treated successfully with sequential red cell and plasma exchange. EJHaem. 2022;4(1):207-210.
doi pubmed pmc - Swerdlow PS. Red cell exchange in sickle cell disease. Hematology Am Soc Hematol Educ Program. 2006;2006(1):48-53.
doi pubmed - Stoltenberg JJ, Gustilo RB. The use of methylprednisolone and hypertonic glucose in the prophylaxis of fat embolism syndrome. Clin Orthop Relat Res. 1979;(143):211-221.
pubmed - Fischer JE, Turner RH, Herndon JH, Riseborough EJ. Massive steroid therapy in severe fat embolism. Surg Gynecol Obstet. 1971;132(4):667-672.
pubmed - Kammeyer R, Devnani R, Mehta R. Cerebral fat embolism syndrome mimicking thrombotic thrombocytopenic purpura in a patient with hemoglobin SC disease. Am J Hematol. 2016;91(5):539-542.
doi pubmed - Myers CF, Ipe TS. Bone Marrow Necrosis in Sickle Cell-Beta Thalassemia Patient Mimicking Thrombotic Thrombocytopenic Purpura. Ann Clin Lab Sci. 2018;48(5):670-673.
pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.