

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://www.thejh.org |
Case Report
Volume 13, Number 4, August 2024, pages 158-163

A Case of Autoimmune Myelofibrosis Associated With Autoimmune Hepatitis
Hailey Tyndalla, d ![]() , Lawrence Worobetza, b, Matthew Nicholsonc, d
, Lawrence Worobetza, b, Matthew Nicholsonc, d
aDepartment of Medicine, University of Saskatchewan, Saskatoon, SK, Canada
bRoyal University Hospital, Saskatoon, SK, Canada
cSaskatoon Cancer Center, Saskatchewan Cancer Agency, and College of Medicine, University of Saskatchewan, Saskatoon, SK, Canada
dCorresponding Author: Hailey Tyndall, Department of Medicine, University of Saskatchewan, Saskatoon, SK, Canada; Matthew Nicholson, Saskatoon Cancer Center, Saskatchewan Cancer Agency, and College of Medicine, University of Saskatchewan, Saskatoon, SK, Canada
Manuscript submitted June 7, 2024, accepted July 15, 2024, published online July 30, 2024
Short title: AIMF and AIH
doi: https://doi.org/10.14740/jh1297
| Abstract | ▴Top |
Autoimmune myelofibrosis (AIMF) is a distinct, underrecognized, and rare cause of bone marrow fibrosis. It carries a favorable outcome and responds well to immunosuppression. Systemic lupus erythematosus is the most common association with AIMF, but there are other cases of associated autoimmune disorders defined in the literature. A 44-year-old female presented to hospital with a 1-month history of fatigue, malaise, and jaundice. She was found to be pancytopenic with elevated liver enzymes. Tests for Janus kinase 2, myeloproliferative leukemia, and calreticulin mutations were negative. Extensive investigations for hemolytic anemia including direct antiglobulin test, flow cytometry for paroxysmal nocturnal hemoglobinuria, testing for hereditary hemoglobinopathies, and hereditary red cell membrane disorders were non-contributory. Antinuclear antibody was positive at > 1,280, immunoglobulin G was 17.04 g/L, and anti-smooth muscle antibody (ASMA) was positive at 1:40. Characteristic features of AIMF on bone marrow biopsy led to the diagnosis of AIMF. The patient was started on prednisone 1 mg/kg with prolonged taper. Fibroscan and liver biopsy were consistent with cirrhosis and workups for other causes of liver dysfunction were unremarkable. She met criteria for diagnosis of autoimmune hepatitis (AIH). The pancytopenia and liver enzymes improved with prednisone. After 1 year of clinical stability, the patient had relapse of disease with pancytopenia, elevated liver enzymes, and similar fibrosis on repeat bone marrow biopsy. Prednisone was reinitiated at 1 mg/kg, and she was started on mycophenolate mofetil (MMF). Prednisone was tapered, and she continues to have an excellent response on MMF alone. We report a case of AIMF associated with AIH, complicated by non-immune hemolysis. AIMF is rare, and its association with AIH is described in only four other cases in the English-language literature. Overlapping biochemical features of AIH and non-immune hemolysis, which has not been well described in AIMF, lead to significant diagnostic complexity and delay. Despite this, a rapid response to corticosteroids was observed including reversal of profound transfusion dependence, normalization of hemoglobin, and reversal of biochemical evidence of hepatic inflammation. A shared pathogenesis of autoimmune fibrosis in both the bone marrow and liver is speculative but suggested by the temporal association in this case.
Keywords: Autoimmune; Bone marrow failure; Myelofibrosis; Hemolysis; Hepatitis; Immunosuppression; Pancytopenia
| Introduction | ▴Top |
Autoimmune myelofibrosis (AIMF) is a distinct and underrecognized cause of bone marrow fibrosis associated with the presence of autoantibodies (primary) or autoimmune disease (secondary) [1]. AIMF is a benign cause of bone marrow fibrosis that carries a favorable outcome and responds well to immunosuppression. This is in contrast to primary myelofibrosis associated with specific mutations such as Janus kinase 2 (JAK2), calreticulin (CALR), or myeloproliferative leukemia (MPL), which has a poor prognosis and the potential to progress to acute myeloid leukemia [2].
Systemic lupus erythematosus (SLE) is the most common condition associated with AIMF, but there are other cases of associated autoimmune disorders defined in the literature. We report a case of secondary AIMF associated with autoimmune hepatitis (AIH). On review of the English-language literature, we found four cases of AIMF associated with AIH [1-4]. This rare association makes this case unique, along with the diagnostic complexity that developed due to the concurrent liver dysfunction and non-immune hemolysis.
| Case Report | ▴Top |
Investigations
A 44-year-old female presented to the emergency department with a 1-month history of fatigue, malaise, and jaundice. Her family physician arranged for blood work, and she was advised to present to the emergency department with pancytopenia. At presentation, she had a past medical history of type II diabetes mellitus and polycystic ovarian syndrome with no known autoimmune history. Family history was unremarkable. She was taking Humalog and rare naproxen for musculoskeletal pain. She did not consume alcohol. Physical examination was unremarkable aside from jaundice.
Lab values showed white blood cell count (WBC) of 2.9 × 109/L, hemoglobin of 92 g/L, mean corpuscular volume (MCV) of 102.7 fL, platelets of 97 × 109/L, reticulocyte count of 68 × 109/L, haptoglobin of < 0.10 g/L, lactate dehydrogenase (LDH) of 386 U/L (reference range 130 - 230 U/L), and total bilirubin of 85 µmol/L. The direct bilirubin was 18 µmol/L and indirect bilirubin was 51 µmol/L. Albumin was 32 g/L, international normalized ratio (INR) was 1.3, aspartate aminotransferase (AST) was 30 U/L, alanine aminotransferase (ALT) was 15 U/L, alkaline phosphatase (ALP) was 81 U/L, and gamma glutamyl transferase (GGT) was 50 U/L. Blood counts from 1 year prior showed moderate thrombocytopenia with hemoglobin of 138 g/L, MCV of 96.0 fL, and platelets of 98 × 109/L. At this point, the patient was identified to have pancytopenia, abnormal liver function tests, unremarkable liver enzymes, and worsening macrocytic anemia including suspected hemolysis confounded by liver dysfunction.
Diagnosis
The direct antiglobulin test (DAT) was negative and peripheral blood smear demonstrated unremarkable red blood cell (RBC) morphology with no schistocytes. Peripheral flow cytometry demonstrated polyclonal lymphocytes, serum protein electrophoresis and serum free light chains revealed polyclonal gammopathy, and vitamin B12 and folate were within normal limits. Further workups including flow cytometry for paroxysmal nocturnal hemoglobinuria, hereditary hemoglobinopathies, RBC membrane disorder genetic panel testing, eosin-5’-maleimide for hereditary red cell membrane disorders, human immunodeficiency virus, hepatitis A/B/C, cytomegalovirus, Epstein-Barr virus, parvovirus, and interferon-gamma release assay were unremarkable. A bone marrow biopsy revealed hypercellular marrow with focal moderate stromal fibrosis with no evidence of lymphoproliferative disorders.
After excluding known hemolytic conditions, the focus turned to the unexplained abnormal liver function tests, raising the possibility of non-alcoholic fatty liver disease, portal hypertension, splenomegaly, and liver cirrhosis. The patient’s abdominal ultrasound showed an unremarkable liver with 18.5 cm splenomegaly. Antinuclear antibody (ANA) was positive at > 1,280, immunoglobulin G (IgG) was elevated at 17.04 g/L and anti-smooth muscle antibody (ASMA) was positive at 1:40. Hepatitis A/B/C testing was negative. The INR remained elevated at 1.3 - 1.4 despite vitamin K supplementation.
The patient’s blood counts remained stable during admission and did not require transfusions or further interventions. The patient was discharged from hospital with outpatient hepatology referral and hematology follow-up with complete blood count (CBC) at least monthly.
The patient represented to the emergency department after 9 months of clinical stability with worsening fatigue, malaise, and dyspnea. Physical examination revealed jaundice and splenomegaly. Her blood work showed WBC of 1.6 × 109/L, hemoglobin of 63 g/L, and platelets of 61 × 109/L. Total bilirubin was 166 µmol/L, albumin was 33 g/L, and INR was 1.5. AST was 45 U/L, ALT was 14 U/L, ALP was 84 U/L, and GGT was 39 U/L. Over the next 5 weeks, she required 12 units packed RBC transfusions to maintain a hemoglobin of > 70 g/L.
A repeat bone marrow biopsy demonstrated increased marrow fibrosis (moderate to focally marked marrow fibrosis, versus prior moderate marrow fibrosis), hypercellularity, erythroid predominance, and increased T-cell predominant lymphoid infiltrates (Fig. 1). Dysplastic changes were absent and cytogenetic studies were normal. Mutations associated with myeloid malignancies across 40 key genes including JAK2, CALR, and MPL were assessed in genomic DNA extracted from the submitted specimen by next-generation sequencing (NGS) using Oncomine Myeloid Research Assay from ThermoFisher and no myeloid mutations were detected. A provisional diagnosis of AIMF was made based on characteristic features on bone marrow biopsy. She did not meet diagnostic criteria for SLE.
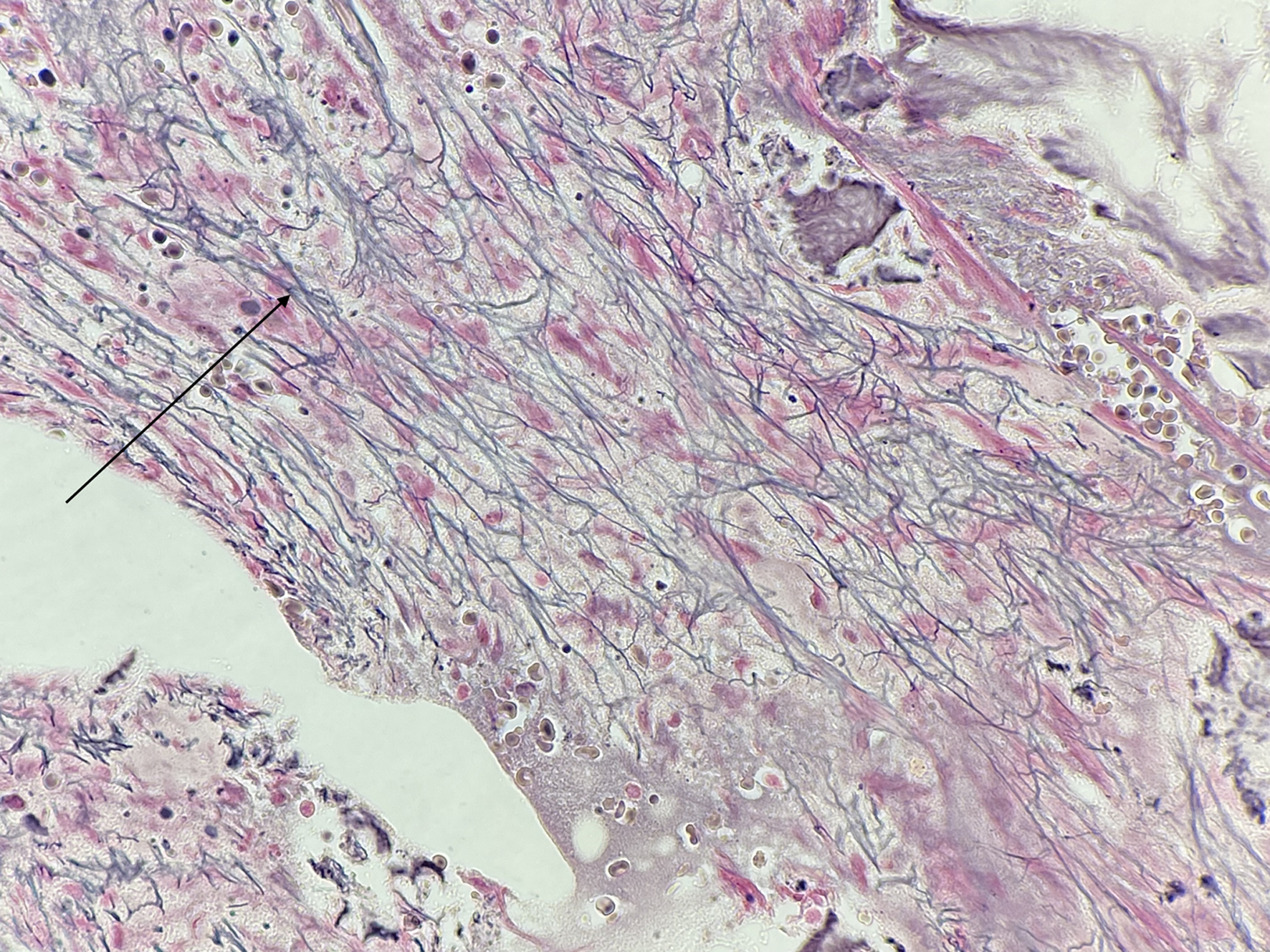 Click for large image | Figure 1. Second bone marrow biopsy with lymphocyte infiltration and fibrosis (arrow). |
Concurrently, the patient was diagnosed with AIH due to high titer ANA, ASMA, elevated IgG, elevated conjugated bilirubin and rising transaminitis. Fibroscan revealed a high fat content with controlled attenuation parameter value 327 dB/m and fibroscan value of 12.8 kPa, in keeping with cirrhosis. Liver biopsy revealed mild chronic inflammation and portal fibrosis (Fig. 2).
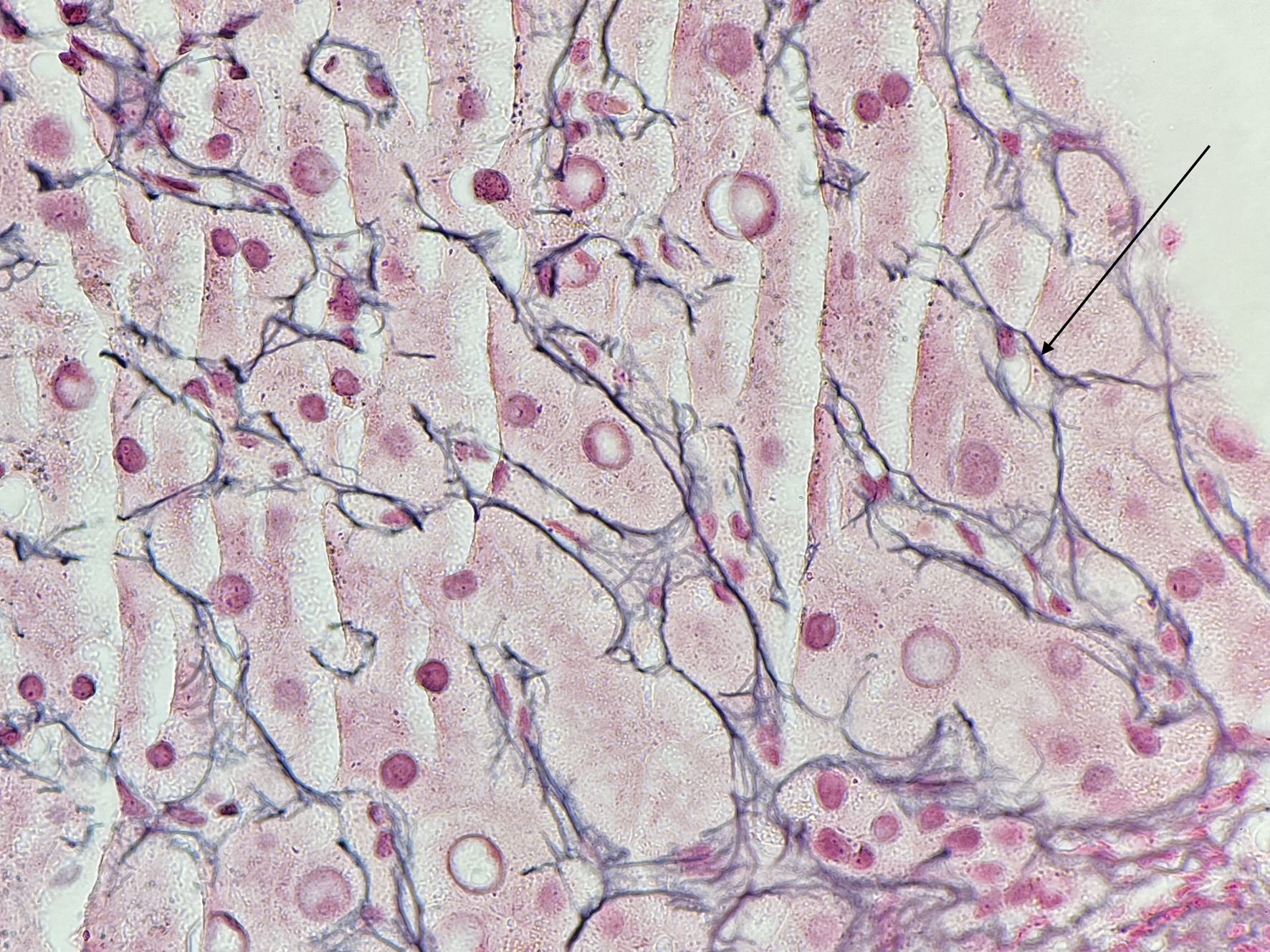 Click for large image | Figure 2. Portal fibrosis (arrow) of the liver. |
Treatment
The patient was treated with 1 mg/kg of prednisone with excellent initial response including rapid de-escalation of transfusion requirements (Fig. 3) with normalization of neutrophil counts and hemoglobin. Serum transaminases and IgG peaked shortly after the initiation of prednisone and returned to normal levels after 2 months of treatment. The prednisone was tapered over 10 weeks. Her hemoglobin, neutrophils, and liver enzymes remained normal for 1 year.
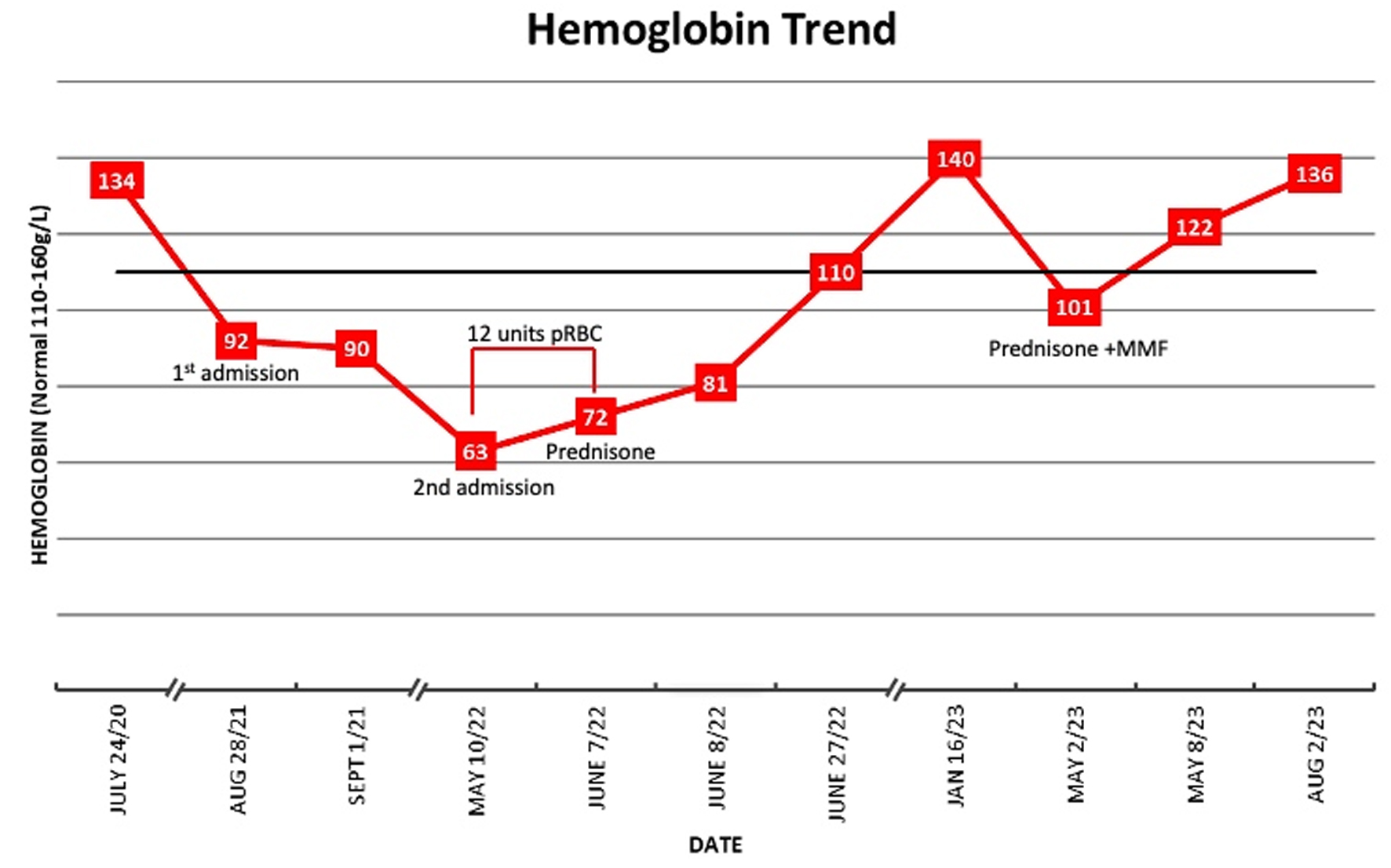 Click for large image | Figure 3. Hemoglobin trend. |
Follow-up and outcomes
After 1 year, the patient developed recurrence with fatigue, jaundice, chills, and malaise. Anemia, reticulocytopenia, polyclonal gammopathy, transaminitis and hyperbilirubinemia recurred, and repeat bone marrow biopsy showed similar fibrosis without any identifiable myeloid mutations. She was treated for relapsed AIMF with prednisone 1 mg/kg and started on mycophenolate mofetil (MMF) 500 mg twice daily. Corticosteroids were rapidly tapered down due to side effects, but response remains excellent on MMF with resolution of anemia with hemoglobin of 142 g/L on her most recent CBC.
| Discussion | ▴Top |
AIMF is a distinct and underrecognized cause of bone marrow fibrosis and lymphocytic infiltration [5]. Proposed diagnostic criteria includes: 1) Grade 3 or 4 bone marrow reticulin fibrosis; 2) Lack of clustered or atypical megakaryocytes; 3) Lack of myeloid or erythroid dysplasia, eosinophilia, or basophilia; 4) Lymphocyte infiltration of the bone marrow; 5) Lack of osteosclerosis; 6) Absent or mild splenomegaly; 7) Presence of autoantibodies; and 8) Absence of a disorder known to cause myelofibrosis [5]. Primary AIMF is associated with the presence of autoantibodies alone, while secondary AIMF is associated with an established autoimmune disease [5]. Majority of cases are associated with SLE, but there are other rare cases of autoimmune disease associations [6, 7]. AIMF is a benign cause of bone marrow fibrosis, in contrast with primary myelofibrosis [2]. It is important to establish the correct diagnosis as these carry different prognoses, treatments, and outcomes [2].
There is significant debate about how to interpret fractionated bilirubin and other markers of hemolysis in the context of liver synthetic dysfunction. In our case, the presence of undetectable haptoglobin, dominant manifestation of severe anemia, and highly elevated indirect bilirubin prompted an extensive workup for acquired and hereditary causes of hemolysis. Autoimmune hemolytic anemia often occurs in association with other autoimmune conditions, but in this case no spherocytes or other morphologic abnormalities were seen and two platforms for DAT, including IgG, complement component 3, immunoglobulin A, and immunoglobulin M were negative. During the period of intensive transfusion support, LDH peaked at 818 U/L, modest elevations in transaminases (AST 45 U/L, ALT normal) and bilirubin peak was 208 µmol/L with indirect bilirubin 186 µmol/L. During this time, reticulocyte counts were inappropriately low at 29 × 109/L (range 19 - 73 × 109/L). Rare syndromes of non-immune hemolysis have been described including intramedullary hemolysis in myelodysplastic syndrome or severe megaloblastic anemia, but a distinct mechanism for non-immune hemolysis has not been described in AIMF or AIH.
AIMF is a rare and underrecognized cause of pancytopenia [5]. This case represents a rare association between AIMF and AIH with concurrent onset and recurrence. This association between AIMF and AIH is described in four other cases in the English-language literature [1-4]. AIMF is rare, and even more rarely associated with AIH in the literature, with majority of cases occurring in those with SLE [6, 7]. Immunosuppression is the treatment for both AIMF and AIH and the same treatment was effective in inducing biochemical remission of both conditions. A shared pathogenesis of autoimmune fibrosis in both marrow and liver is speculative but is suggested by the temporal association in this case.
Clinicians should consider an autoimmune workup in unexplained cytopenias after ruling out the usual offenders, especially in young or middle-aged populations, women, or those with a history of autoimmune disease. It is important to distinguish AIMF from primary myelofibrosis, which carries a different treatment approach and prognosis [2]. In primary myelofibrosis, the foremost concerns are thrombotic events, cytopenias, and progression to acute myeloid leukemia [8, 9]. In contrast, the prognosis of AIMF is favorable if it is diagnosed and treated appropriately, including reversibility of marrow fibrosis in many cases [5].
Whether corticosteroids should be used alone or in combination with a steroid sparing agent up front is not well established. In those with SLE or other associated autoimmune diseases, AIMF may require long-term immunosuppression, or multiple immunosuppressive agents, to achieve sustained remission [2, 10]. In this case, the patient was treated with corticosteroid monotherapy and the result was rapid clinical and laboratory remission, but with only 1 year until clear relapse. Our experience likely supports prior expert recommendations to institute corticosteroid-sparing agents up front in those with secondary AIMF.
This is a case of secondary AIMF associated with AIH. We observed dramatic response to treatment in this young woman requiring 12 units of packed RBCs over 5 weeks to maintaining a normal hemoglobin level with immunosuppression. Establishing the correct diagnosis and initiating immunosuppression in AIMF remain significant challenges, and clinical awareness of this condition in complex cases of cytopenias, particularly those with other autoimmune conditions, is critical.
Learning points
AIMF is a rare cause of pancytopenia that is usually only considered in patients with SLE. It is important to consider this diagnosis in those presenting with cytopenias who are younger in age, women, or those with known autoimmune diseases. Non-immune hemolysis may be a presenting feature of AIMF. It is critical to distinguish AIMF from primary myelofibrosis as they carry different prognoses, outcomes, and treatment. Myeloid mutations are present in primary myelofibrosis and absent in AIMF. In AIMF, corticosteroid treatment is generally effective, but relapse is common and combination with corticosteroid-sparing agents should be considered, particularly in cases of secondary AIMF.
Acknowledgments
We acknowledge and thank the patient for her involvement.
Financial Disclosure
There are no funding or financial disclosures to report for this submission.
Conflict of Interest
There is no conflict of interest to disclose.
Informed Consent
This study was deemed exempt from the requirement of Research Ethics Board review. Informed, written consent was provided by the patient for the study participation and for publication.
Author Contributions
LW and MN contributed to the identification and management of the case. HT, LW, and MN wrote and reviewed the manuscript.
Data Availability
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Abbreviations
AIH: autoimmune hepatitis; AIMF: autoimmune myelofibrosis; ALP: alkaline phosphatase; ALT: alanine transaminase; ANA: antinuclear antibody; ASMA: anti-smooth muscle antibody; AST: aspartate aminotransferase; CALR: calreticulin; CBC: complete blood count; DAT: direct antiglobulin test; GGT: gamma-glutamyl transferase; IgG: immunoglobulin G; INR: international normalized ratio; JAK2: Janus kinase 2; LDH: lactate dehydrogenase; MCV: mean corpuscular volume; MMF: mycophenolate mofetil; MPL: myeloproliferative leukemia; RBC: red blood cell; SLE: systemic lupus erythematosus; WBC: white blood cell
| References | ▴Top |
- Ohkawara H, Furukawa M, Ikeda K, Shichishima-Nakamura A, Fukatsu M, Sano T, Ueda K, et al. Steroid-resistant autoimmune myelofibrosis in a patient with autoimmune hepatitis and Evans syndrome complicated with increased expression of TGF-beta in the bone marrow: a case report. Int J Hematol. 2017;106(5):718-724.
doi pubmed - Vergara-Lluri ME, Piatek CI, Pullarkat V, Siddiqi IN, O'Connell C, Feinstein DI, Brynes RK. Autoimmune myelofibrosis: an update on morphologic features in 29 cases and review of the literature. Hum Pathol. 2014;45(11):2183-2191.
doi pubmed - Gruson B, Brevet M, Vaida I, Sid idris S, Damaj G. Myelofibrosis and cytopenia are not always malignant. Eur J Intern Med. 2006;17(2):136-137.
doi pubmed - Gutarra-Arana M, Haider T, Kieffer T, Hugenberg S. Autoimmune myelofibrosis. A case review. The Rheumatologist. 2014. https://www.the-rheumatologist.org/article/autoimmune-myelofibrosis-a-case-review/.
- Pullarkat V, Bass RD, Gong JZ, Feinstein DI, Brynes RK. Primary autoimmune myelofibrosis: definition of a distinct clinicopathologic syndrome. Am J Hematol. 2003;72(1):8-12.
doi pubmed - Bass RD, Pullarkat V, Feinstein DI, Kaul A, Winberg CD, Brynes RK. Pathology of autoimmune myelofibrosis. A report of three cases and a review of the literature. Am J Clin Pathol. 2001;116(2):211-216.
doi pubmed - Prakash S, Alhariri S, Hassan M, Patel PK, Corral J. Improvement in primary autoimmune myelofibrosis following a short course of steroids and intravenous immunoglobulins. Cureus. 2022;14(9):e29735.
doi pubmed pmc - Tefferi A. Myelofibrosis with myeloid metaplasia. N Engl J Med. 2000;342(17):1255-1265.
doi pubmed - Tefferi A. Primary myelofibrosis: 2021 update on diagnosis, risk-stratification and management. Am J Hematol. 2021;96(1):145-162.
doi pubmed - Ramakrishna R, Kyle PW, Day PJ, Manoharan A. Evans' syndrome, myelofibrosis and systemic lupus erythematosus: role of procollagens in myelofibrosis. Pathology. 1995;27(3):255-259.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.