

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website http://www.thejh.org |
Case Report
Volume 3, Number 4, December 2014, pages 112-115
Concurrent Presentation of High-Grade Lymphoma and Metastatic Pancreatic Neuroendocrine Tumor 14 Years After Renal Transplant
Trisha Wise-Drapera, d, Julianne Qualtierib, Gautham Mogilishettyc, Tahir Latifa
aDepartment of Internal Medicine, Division of Hematology/Oncology, University of Cincinnati College of Medicine, Cincinnati, OH 45267, USA
bDepartment of Pathology, University of Cincinnati College of Medicine, Cincinnati, OH 45267, USA
cDepartment of Surgery, Division of Transplantation, University of Cincinnati College of Medicine, Cincinnati, OH 45267, USA
dCorresponding Author: Trisha Wise-Draper, University of Cincinnati, The Vontz Center for Molecular Studies, 3125 Eden Avenue, Cincinnati, OH 45267, USA
Manuscript accepted for publication November 25, 2014
Short title: Concurrent Lymphoma and PNET After Transplant
doi: http://dx.doi.org/10.14740/jh180w
| Abstract | ▴Top |
The development of malignancy, especially lymphoma, is common after solid organ transplant. However, concurrent malignancies are rare and result in a diagnostic and treatment dilemma, particularly in the post-transplant setting. We present a case of a 78-year-old male who was discovered to have a high-grade post-transplant lymphoproliferative disorder (PTLD) and metastatic pancreatic neuroendocrine tumor (PNET) 14 years after kidney transplant. He presented with abdominal pain and at surgical resection was found to have a large small intestine tumor that was consistent with high-grade diffuse large B cell lymphoma. Immune suppression was reduced, and staging workup was completed. Positron emission tomography-computed tomography (PET-CT) showed fluorodeoxyglucose (FDG)-avid metastatic lesions in the liver and a mass in the pancreatic head. Before treatment was initiated, biopsy of a liver lesion revealed metastatic PNET. Due to aggressiveness and potential high mortality of the lymphoma, he was started on rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone (R-CHOP). After five cycles he developed worsening abdominal pain consistent with progression of the PNET and was placed on everolimus. Here we discuss the complexity of diagnosing concurrent primaries and the treatment of such in the post-transplant setting.
Keywords: Cancer; Post-transplant; PNET; PTLD
| Introduction | ▴Top |
Here we present an unusual case of two malignancies, high-grade post-transplant lymphoproliferative disorder (PTLD) and metastatic pancreatic neuroendocrine tumor (PNET), presenting concurrently in the post-kidney transplant setting creating a significant diagnostic and treatment dilemma. The incidence of PTLD has been reported as high as 1% in the first 5 years and 2.1% 10 years after kidney transplantation in a French study [1]. Incidence varies depending on type of prior allograft where heart and lung transplants often carry the greatest risk that is often attributed to the high amount of immunosuppression needed in these patients. Although PTLDs are relatively common after transplant, the incidence of PNETs after solid tumor transplant is not well understood. PNETs are rare tumors with an incidence of 1/100,000 per year in the general population and accounting for only 1-2% of pancreatic tumors [2]. Additionally, it is unclear if there is an association with the occurrence of PNET and prior solid organ transplant.
| Case Report | ▴Top |
A 78-year-old African-American male recipient of a renal transplant 14 years prior on immunosuppressive therapy presented with a 2-day history of abdominal pain and nausea without emesis. His pain was similar to a prior episode in which he had volvulus and obstruction requiring surgical intervention. Abdominal X-ray was concerning for obstruction, and CT scan showed small bowel obstruction as well as hepatic lesions which were poorly characterized. Exploratory laparotomy revealed a palpable mass in the terminal ileum. He underwent ileocecectomy with resection of the mass. The mass was described as an ulcerated tumor composed of sheets of discohesive, randomly oriented cells which appear lymphoid in origin with enlarged irregular convoluted nuclei with scant eosinophilic cytoplasm. Relatively numerous mitotic figures were seen. The infiltrate extended through the muscular wall into serosal tissue. Immunohistochemical staining showed that the cells were positive for cluster of differentiation 45 (CD45), CD20, CD10 and bcl-6. The tumor was additionally negative for CD34, terminal deoxynucleotidyl transferase (TdT), CD117, CD30, anaplastic lymphoma kinase (ALK-1), epithelial membrane antigen (EMA) and myeloperoxidase as well as Epstein-Barr encoded RNA (EBER) although a qualitative Epstein-Barr virus (EBV) polymerase chain reaction (PCR) was positive on peripheral blood. The pathology was consistent with a high-grade B cell lymphoma described as a diffuse large B cell lymphoma of germinal center origin (Fig. 1A, B). Approximately four of the identified mesenteric lymph nodes showed partial involvement by the lymphoma. No perforation of the bowel was identified. Other laboratory tests revealed an elevated lactate dehydrogenase of 679 U/L, hemoglobin of 11.2 g/dL with normal white blood cell count and platelets. Creatinine was slightly elevated at 1.3 mg/dL which was similar to his baseline and alkaline phosphatase was also slightly elevated in the 140s (U/L). Due to concern for post-transplant lymphoma (PTLD), his immunosuppression was decreased and prednisone was added to his regimen.
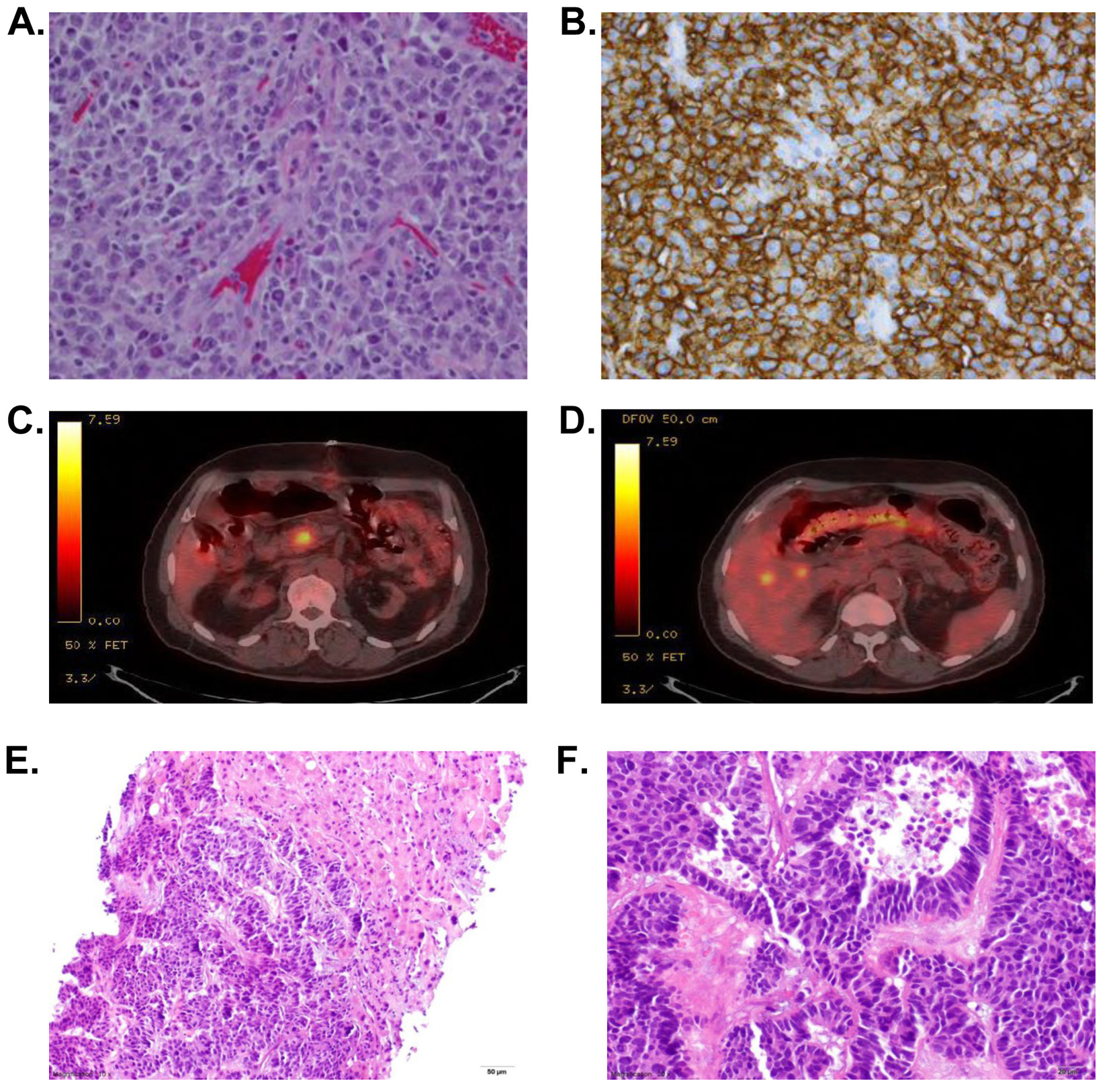 Figure 1. (A, B) Diffuse large B cell lymphoma. (A) Hematoxylin and eosin (H&E) stain at × 40 of resected tumor from terminal ileum showing sheets of large, pleomorphic cells with prominent nucleoli. (B) CD20 stain which was highly positive confirming this tumor to be of B cell origin. (C, D) Representative frames of the PET-CT demonstrating FDG uptake in liver and pancreatic head after resection of small bowel tumor. (E, F): PNET. Representative H&E stains from biopsy of a liver tumor from our patient demonstrating a nested and trabecular arrangement of neoplastic cells with high N:C ratios, frequent mitoses, focal necrosis and uniform chromatin at × 10 (E) and × 20 (F). Figure 1. (A, B) Diffuse large B cell lymphoma. (A) Hematoxylin and eosin (H&E) stain at × 40 of resected tumor from terminal ileum showing sheets of large, pleomorphic cells with prominent nucleoli. (B) CD20 stain which was highly positive confirming this tumor to be of B cell origin. (C, D) Representative frames of the PET-CT demonstrating FDG uptake in liver and pancreatic head after resection of small bowel tumor. (E, F): PNET. Representative H&E stains from biopsy of a liver tumor from our patient demonstrating a nested and trabecular arrangement of neoplastic cells with high N:C ratios, frequent mitoses, focal necrosis and uniform chromatin at × 10 (E) and × 20 (F). |
A PET-CT (Fig. 1C, D) was performed as well as bone marrow biopsy to complete the workup. Bone marrow biopsy showed no involvement of lymphoma. His PET-CT showed focal fluorodeoxyglucose (FDG) uptake in the pancreatic head and multiple hypodense liver lesions but no other lymphadenopathy. He underwent a liver biopsy to characterize these atypical liver lesions. His liver biopsy showed grade II metastatic pancreatic neuroendocrine carcinoma (PNET) confirming two synchronous primary cancers (Fig. 1E, F). The liver tumor was composed of medium-to-large cells with frequent mitosis (up to five per high power field) and focal necrosis. The tumor cells were diffusely positive for neuroendocrine marker synaptophysin and chromogranin and negative for lung marker thyroid transcription factor (TTF-1), squamous marker p63, colonic marker CK20, and hepatocytic markers hepatocyte paraffin 1 and glypican-3. The tumor cells were strongly positive for CK7 and weakly positive for CDX2. Due to extensive local involvement and concern for recurrence of lymphoma, he commenced standard therapy with rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone (R-CHOP) after immune suppression had been decreased. The patient was able to complete five cycles of R-CHOP with no sign of recurrence of lymphoma but unfortunately had progression of the PNET. He was started on 5 mg of everolimis daily; however, he was unable to tolerate the medication and therefore, pursued palliative care.
| Discussion | ▴Top |
As mentioned above, PTLD incidence varies greatly with the type of allograft but can reach as high as 5% cumulative incidence especially after heart/lung transplantation. The risk of non-Hodgkin’s lymphoma has been reported to be greater than 20 fold higher after kidney transplant compared to the general population, and the incidence is cumulative in those with a solid organ transplant [3, 4]. More importantly, the prognosis of PTLD is poor with only 59% overall survival (OS) at 5 years especially in those that develop PTLD early after transplantation [5]. A more recent study demonstrated a mortality rate of 26% in those after kidney transplant in the setting of new therapy options [6]. Current treatment is primarily chosen based on aggressiveness of disease and prognostic factors. Initially withdraw/reduction of immunosuppression is often performed which can result in 40% regression. The latter is thought to be mediated by allowing the natural immune system to regain control of EBV infection, but this effect is often limited to more indolent disease. Additionally, there must be a balance between treatment of the lymphoma and rejection of the allograft. In aggressive disease, as in our patient based on increased mitotic figures, targeted therapy and chemotherapy are often added. Rituximab alone can achieve response rates of 50% but are often associated with relapse [7, 8]. A phase II trial showed that the addition of CHOP after rituximab resulted in 90% complete response rates in adult patients who had failed initial withdraw of immunosuppression; however, the toxicity was high at 11% [9]. Although there is no current standard, in patients with high-risk monoclonal disease, it is accepted to add chemotherapy to achieve a long-lasting response and prevent relapse.
Although, there is a known increased risk of non-Hodgkin’s lymphoma in the post-transplant setting, the incidence of PNETs after solid tumor transplant is unknown. In a large cohort study that linked the United States scientific registry transplant recipients from 1987 to 2008 and 13 state cancer registries, investigators compared standardized incident ratios (SIRs) in 175,732 solid transplant recipients which demonstrated that there was an increased SIR of pancreatic cancers of 1.46 [10]. However, it is unclear whether this increased risk included PENTs. Immune suppression and viral infection have been linked as possible causes of many malignancies after transplant, although this does not account for all cases. There have been small reports that immune suppression itself may play a role in the development of neuroendocrine tumors as in human immunodeficiency virus (HIV) infection and in the post-transplant setting (cancer.net) [11], but as of yet, there has not been a clear association with solid organ transplant and the development of PNETs. The incidence in the general population as mentioned above is approximately 1/100,000 and accounts for a small number of all pancreatic cancers. For patients with localized disease and/or localized metastatic involvement of the liver, resection can be offered for curative intent. However, this is not an option in patients with poor hepatic function or bilobar involvement of the liver. In asymptomatic metastatic PNET, observation is acceptable until symptoms develop. For symptomatic PNET, somatastatin analogs, everolimus and sunitinib are all options for first-line treatment. Chemotherapy is often reserved for large tumor bulk or poorly differentiated PNET requiring quick initial response.
This case is unique in that the patient developed two primary cancers after renal transplant. Concurrent primaries often provide a dilemma for diagnosis and treatment, and in this case it was even more complicated by history of renal transplant. The liver lesions were initially thought to be related to lymphoma, but based on suspicion from location and appearance, we were concerned about a second cancer. Therefore, biopsy was pursued which was important in this case to determine treatment and response. As mentioned above, the two tumors are treated very differently. Both tumors were felt to be relatively aggressive but the PTLD was felt to be the primary concern due to early high mortality if not treated. Due to extensive local involvement, aggressiveness of pathological features and concern for recurrence, he commenced therapy with R-CHOP after immune suppression had been decreased. Doxorubicin has been shown to have activity in PNET in combination with streptozocin [12] and therefore, in this case, was felt to potentially target both tumors. The patient was able to complete five cycles of R-CHOP but unfortunately had progression of the liver lesions with severe abdominal pain. Based on the previous biopsy of the liver, we concluded that his lymphoma was in remission but that the PNET was progressing. He commenced everolimus as first-line PNET treatment but did not tolerate therapy well and opted for palliative care.
Currently, there is not sufficient data to determine if PNET is more common after transplant and if these tumors have different clinical behaviors. It is important to have clinical suspicion for the development of other rare tumor types after transplant, as synchronous primaries can occur as in this case and require specialized treatment and attention.
Acknowledgement
We would like to thank Craig Isenhart from the Jewish Hospital in Cincinnati, Ohio for providing pictures of the patient’s lymphoma.
Conflict of Interest
The authors of this manuscript have no conflicts of interest to disclose.
Abbreviations
PTLD: post-transplant lymphoproliferative disorder; PNET: pancreatic neuroendocrine tumor; PET-CT: positron emission tomography-computed tomography; FDG: fluorodeoxyglucose; R-CHOP: rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone; CD: cluster of differentiation; TdT: terminal deoxynucleotidyl transferase; ALK-1: anaplastic lymphoma kinase 1; EMA: epithelial membrane antigen; EBER: Epstein-Barr encoded RNA; EBV: Epstein-Barr virus; PCR: polymerase chain reaction; TTF-1: thyroid transcription factor; SIR: standardized incident ratios; HIV: human immunodeficiency virus
| References | ▴Top |
- Caillard S, Lamy FX, Quelen C, Dantal J, Lebranchu Y, Lang P, Velten M, et al. Epidemiology of posttransplant lymphoproliferative disorders in adult kidney and kidney pancreas recipients: report of the French registry and analysis of subgroups of lymphomas. Am J Transplant. 2012;12(3):682-693.
doi pubmed - Kloppel G, Perren A, Heitz PU. The gastroenteropancreatic neuroendocrine cell system and its tumors: the WHO classification. Ann N Y Acad Sci. 2004;1014:13-27.
doi pubmed - Kasiske BL, Snyder JJ, Gilbertson DT, Wang C. Cancer after kidney transplantation in the United States. Am J Transplant. 2004;4(6):905-913.
doi pubmed - Hall EC, Pfeiffer RM, Segev DL, Engels EA. Cumulative incidence of cancer after solid organ transplantation. Cancer. 2013;119(12):2300-2308.
doi pubmed - Hauke R, Smir B, Greiner T, Bierman P, Tarantolo S, Anderson J, Shaw B, et al. Clinical and pathological features of posttransplant lymphoproliferative disorders: influence on survival and response to treatment. Ann Oncol. 2001;12(6):831-834.
doi pubmed - Wasson S, Zafar MN, Best J, Reddy HK. Post-transplantation lymphoproliferative disorder in heart and kidney transplant patients: a single-center experience. J Cardiovasc Pharmacol Ther. 2006;11(1):77-83.
doi pubmed - Choquet S, Leblond V, Herbrecht R, Socie G, Stoppa AM, Vandenberghe P, Fischer A, et al. Efficacy and safety of rituximab in B-cell post-transplantation lymphoproliferative disorders: results of a prospective multicenter phase 2 study. Blood. 2006;107(8):3053-3057.
doi pubmed - Choquet S, Oertel S, LeBlond V, Riess H, Varoqueaux N, Dorken B, Trappe R. Rituximab in the management of post-transplantation lymphoproliferative disorder after solid organ transplantation: proceed with caution. Ann Hematol. 2007;86(8):599-607.
doi pubmed - Trappe R, Oertel S, Leblond V, Mollee P, Sender M, Reinke P, Neuhaus R, et al. Sequential treatment with rituximab followed by CHOP chemotherapy in adult B-cell post-transplant lymphoproliferative disorder (PTLD): the prospective international multicentre phase 2 PTLD-1 trial. Lancet Oncol. 2012;13(2):196-206.
doi - Engels EA, Pfeiffer RM, Fraumeni JF, Jr., Kasiske BL, Israni AK, Snyder JJ, Wolfe RA, et al. Spectrum of cancer risk among US solid organ transplant recipients. JAMA. 2011;306(17):1891-1901.
doi pubmed - Lito P, Pantanowitz L, Marotti J, Aboulafia DM, Campbell V, Bower M, Dezube BJ. Gastroenteropancreatic neuroendocrine tumors in patients with HIV infection: a trans-Atlantic series. Am J Med Sci. 2009;337(1):1-4.
doi pubmed - Moertel CG, Lefkopoulo M, Lipsitz S, Hahn RG, Klaassen D. Streptozocin-doxorubicin, streptozocin-fluorouracil or chlorozotocin in the treatment of advanced islet-cell carcinoma. N Engl J Med. 1992;326(8):519-523.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.