

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website http://www.thejh.org |
Case Report
Volume 4, Number 3, September 2015, pages 193-195
An Atypical Case of Atypical Hemolytic Uremic Syndrome: Predominant Gastrointestinal Involvement, Intact Renal Function, and C5b-9 Deposition in Colon and Skin
Jocelyn De Yaoa, Robert Kaplana, c, Cynthia Magrob
aWestern Pennsylvania Cancer Institute, Allegheny Health Network, 4800 Friendship Ave, Ste 2303, Pittsburgh, PA 15224, USA
bDepartment of Dermatopathology, Weill Cornell Medical College, 525 East 68th St, Room F-309, New York, NY 10065, USA
cCorresponding Author: Robert Kaplan, Western Pennsylvania Cancer Institute, Allegheny Health Network, 4800 Friendship Ave, Ste 2303, Pittsburgh, PA 15224, USA
Manuscript accepted for publication July 19, 2015
Short title: Atypical Hemolytic Uremic Syndrome
doi: http://dx.doi.org/10.14740/jh205w
| Abstract | ▴Top |
Atypical hemolytic uremic syndrome (aHUS) is one of the prototypic thrombotic microangiopathies which arises from a genetically-based defect in the regulatory control of the alternate complement cascade. Although its cause is distinct, it shares a similar clinical presentation with thrombotic thrombocytopenic purpura with respect to its pattern of organ involvement, with most cases including the renal, central nervous, and gastrointestinal systems. Renal dysfunction in aHUS is generally recognized as a clinical hallmark of the disease. We report a unique case of aHUS in a 71-year-old female highlighted by predominant effect on the gastrointestinal system concurrent with preservation of renal function and histopathologically-proven involvement of skin.
Keywords: aHUS; Thrombotic microangiopathy
| Introduction | ▴Top |
Thrombotic microangiopathy (TMA), a systemic disorder affecting the microvasculature, is characterized by microangiopathic hemolytic anemia, consumptive thrombocytopenia, and vascular thrombosis. Atypical hemolytic uremic syndrome (aHUS) is a previously under-recognized form of TMA which, in the majority of cases is a consequence of a genetically based dysregulation of the alternate complement pathway (AP) [1-5]. Microangiopathy in aHUS is the end-organ effect of a systemic complement-mediated diathesis which has the propensity to affect the vascular bed of all organs but exhibits a particular affinity for the renal, central nervous, and gastrointestinal systems (in this order of frequency). Historically, the majority of reported cases have included some degree of renal dysfunction, frequently including end-stage renal disease. Skin involvement is not a commonly reported finding, but both clinically apparent and sub-clinical involvements have been described in some recent publications [6, 7]. We describe, herein, a unique case of aHUS highlighted by recurrent pancreatitis, ischemic colitis, and preserved renal function, with pathological assessment demonstrating microvascular thrombosis and endothelial complement deposition in colon and skin.
| Case Report | ▴Top |
A 71-year-old Caucasian female (non-drinker) presented to our institution with symptomatic pancreatitis, non-immune hemolytic anemia and thrombocytopenia within 24 h of fine-needle aspiration of a pancreatic cyst. A review of her records revealed a several month history of unexplained biochemical pancreatitis. She was initially managed conservatively with IV hydration and opioid analgesia, but her course was later complicated by pancreatic hemorrhage. CT imaging failed to detect biliary or pancreatic duct dilatation while the gall-bladder was surgically absent. Laboratory investigation revealed hyperlipasemia without hyperlipidemia, normocytic anemia with a negative direct Coomb’s, mild thrombocytopenia, and a normal eGFR (Table 1). Further testing detected markers of intravascular hemolysis, normal ADAMTS-13 activity (75%), a reduction in the plasma level of C3 (preserved levels of CH4), normal serologies (including antiphospholipid antibodies), and the presence on peripheral smear of 3 - 5 schistocytes per HPF. With these findings, consideration was given to aHUS as the unifying diagnosis. Without apparent kidney dysfunction, histological evidence of non-renal, TMA-related organ involvement was sought. Attention was focused on sampling easily accessible cutaneous vessels with punch biopsies of non-lesional skin. Light microscopy here revealed features of endothelial injury as well as multiple platelet/fibrin microthrombi. Immunohistochemical assessment with direct immunofluorescence (DIF) detected prominent endothelial deposition of C5b-9 within the cutaneous microvasculature (Fig. 1A-C).
 Click to view | Table 1. Laboratory Findings Pre- and Post-Treatment With Eculizumab |
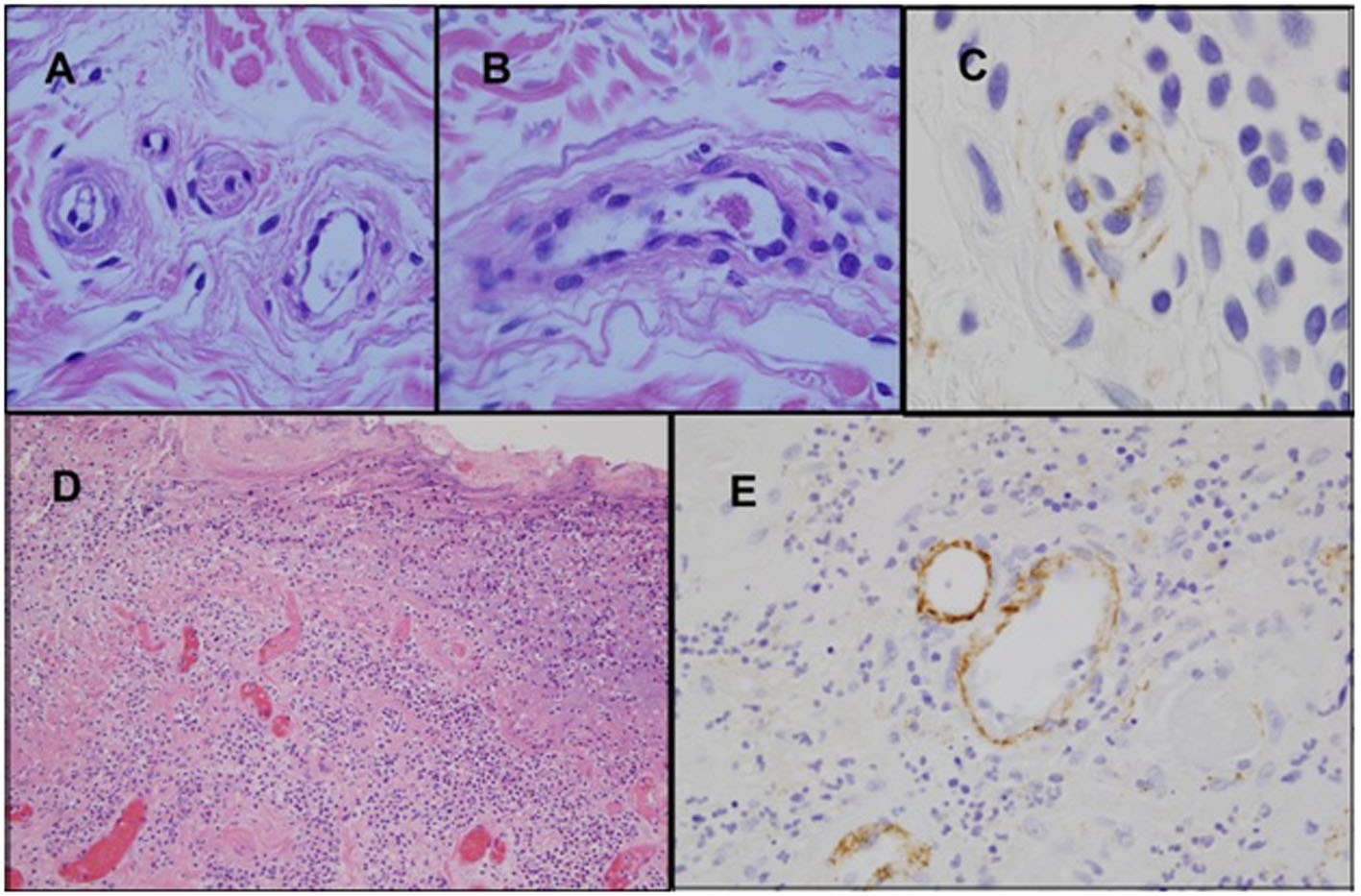 Click for large image | Figure 1. Biopsy of skin and colon. (A-C) Biopsy of non-lesional skin shows endothelial cell detachment with edema of the vessel wall, and concomitant vascular basement membrane zone reduplication (A). A venule contains a small intraluminal thrombus comprised of fibrin and platelets (B) (H&E). Immuohistochemical studies show prominent deposits of C5b-9 within the vessel walls (C) (iaminobenzidene). (D-E) Sections of the colon which demonstrate ischemic ulceration associated with vascular thrombosis (D) (H&E). C5b-9 deposits are prominent within the microvasculature of the colonic mucosa (E) (diaminobenzidene). |
Terminal-complement blockade therapy with the humanized monoclonal anti-Cs antibody, eculizumab, was then initiated. The patient received two weekly doses before treatment was interrupted by feeding tube complications. In the interim, she developed recurrent bouts of pancreatitis with pseudocyst enlargement and clinical suspicion of ischemic bowel. Emergent surgical exploration revealed partial pancreatic necrosis, perforation of the terminal ileum, and an adjacent section of ischemic colon (resected). Pathological evaluation revealed microvascular thrombosis with DIF detecting extensive C5b-9 deposition in the colonic microvasculature (Fig. 1D, E). The patient soon exhibited robust laboratory and clinical improvement after completing 4 weeks of re-treatment with the eculizumab induction schedule. Currently, she remains on the maintenance schedule after having achieved complete resolution of gastrointestinal symptoms and normalization of all laboratory parameters (Table 1).
| Discussion | ▴Top |
As integral components of our immune system, the three arms of the complement cascade possess the capacity for dramatic effects on both self and non-self targets, and thus require meticulous regulatory controls. The alternate pathway is unique in that it is endowed with a constitutive, up-tick mechanism which primes it for rapid amplification and enables it to trigger a sequence of inter-dependent reactions culminating in powerful responses to protect the host. When the system loses its built-in regulatory controls, persistent generation of the terminal-effector proteins (C5a, C5b-9) can subject the host to repeated endothelial insults [8, 9], potentially leading to ischemic injury. Precise control is conferred by a series of regulatory proteins which inhibit activation by blocking specific components of the complement cascade. Inherited mutations in these regulatory proteins render patients exquisitely susceptible to complement-amplification levels beyond their inherent ability to control. The situations known to have complement-amplifying properties are not especially rare events for either ill or healthy patients; they include infectious/inflammatory processes, surgeries and invasive procedures, pregnancy, and autoimmunity [1, 10-13]. In placing our present case into this pathophysiological framework, one can speculate that repeated FNA passes into an organ with pre-existing inflammation in the context of a dysregulated AP would constitute a set of conditions capable of producing ischemic organ destruction.
Clinicians often face considerable diagnostic challenges when encountering patients with suspected aHUS. This is, at least in part, due to the similarity of its clinical presentation to TTP/typical HUS, as well as lack of a confirmatory diagnostic test. Moreover, as a consequence of the ability of the disease to affect the endothelium of any organ, there is significant inter-patient heterogeneity in aHUS regarding patterns of organ involvement and associated symptoms. Our patient met the diagnostic criteria of 1) microangiopathic hemolytic anemia, 2) 25% reduction from baseline platelet count, 3) clinical suspicion of pancreatic involvement by exclusion of other causes, and 4) absence of severe ADAMTS-13 deficiency (< 5%). While acknowledging the potential non-TMA causes for the platelet count decline, an alternate factor for the thrombocytopenia would not invalidate the diagnosis, as it has been observed that approximately 13% of aHUS patients may lack thrombocytopenia at initial presentation [14]. Regarding the criterion of aHUS-related organ involvement, the patient had unexplained chemical pancreatitis ongoing for several months. The absence of any of the common etiologies of pancreatitis as well as the severity and unrelenting nature of its course (uncharacteristic of simple iatrogenic pancreatitis) led us by exclusion to suspect aHUS. The lack of renal dysfunction was viewed as an atypical feature, but not necessarily a finding which would exclude aHUS, as it is estimated that approximately 20% of aHUS patients will have preserved renal function at diagnosis. Prior to the patient developing ischemic bowel, we attempted to obtain objective evidence of TMA-related organ involvement from an easily accessible organ, the integument. Punch biopsies were performed on normal skin based on previous reports of potentially diagnostically useful information being gleaned from histopathological evaluation of cutaneous vessels [6, 7]. Our biopsy results were interpreted as endothelial injury in the presence of platelet/fibrin microthrombi consistent with TMA. In addition, immunohistochemical studies showed prominent deposits of C5b-9 within vessel walls, implicating an underlying complement-associated process. Similar abnormalities were found but with greater intensity when the same studies were later performed on the segment of resected colon (Fig. 1), thereby linking TMA to the pathogenesis of the colonic ischemia as well.
In summary, we report this atypical case of aHUS as being illustrative of the disorder’s potential for varied and distinct clinical presentations, encompassing both clinical and sub-clinical levels of disease. Awareness of this disorder’s broad diversity of clinical scenarios combined with rational use of the available testing should enhance our diagnostic efficiency, thereby optimizing the time-dependent benefit attributed to the emerging array of complement-based therapies.
| References | ▴Top |
- Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361(17):1676-1687.
doi pubmed - Caprioli J, Noris M, Brioschi S, Pianetti G, Castelletti F, Bettinaglio P, Mele C, et al. Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood. 2006;108(4):1267-1279.
doi pubmed - Fremeaux-Bacchi V, Miller EC, Liszewski MK, Strain L, Blouin J, Brown AL, Moghal N, et al. Mutations in complement C3 predispose to development of atypical hemolytic uremic syndrome. Blood. 2008;112(13):4948-4952.
doi pubmed - Maga TK, Nishimura CJ, Weaver AE, Frees KL, Smith RJ. Mutations in alternative pathway complement proteins in American patients with atypical hemolytic uremic syndrome. Hum Mutat. 2010;31(6):E1445-1460.
doi pubmed - Noris M, Ruggenenti P, Perna A, Orisio S, Caprioli J, Skerka C, Vasile B, et al. Hypocomplementemia discloses genetic predisposition to hemolytic uremic syndrome and thrombotic thrombocytopenic purpura: role of factor H abnormalities. Italian Registry of Familial and Recurrent Hemolytic Uremic Syndrome/Thrombotic Thrombocytopenic Purpura. J Am Soc Nephrol. 1999;10(2):281-293.
pubmed - Ardissino G, Tel F, Testa S, Marzano AV, Lazzari R, Salardi S, Edefonti A. Skin involvement in atypical hemolytic uremic syndrome. Am J Kidney Dis. 2014;63(4):652-655.
doi pubmed - Chapin J, Weksler B, Magro C, Laurence J. Eculizumab in the treatment of refractory idiopathic thrombotic thrombocytopenic purpura. Br J Haematol. 2012;157(6):772-774.
doi pubmed - Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344(14):1058-1066.
pubmed - Laurence J. Atypical hemolytic uremic syndrome (aHUS): making the diagnosis. Clin Adv Hematol Oncol. 2012;10(10 Suppl 17):1-12.
pubmed - Sanchez-Corral P, Melgosa M. Advances in understanding the aetiology of atypical Haemolytic Uraemic Syndrome. Br J Haematol. 2010;150(5):529-542.
doi pubmed - Besbas N, Karpman D, Landau D, Loirat C, Proesmans W, Remuzzi G, Rizzoni G, et al. A classification of hemolytic uremic syndrome and thrombotic thrombocytopenic purpura and related disorders. Kidney Int. 2006;70(3):423-431.
doi - Zakarija A, Bennett C. Drug-induced thrombotic microangiopathy. Semin Thromb Hemost. 2005;31(6):681-690.
doi pubmed - Noris M, Caprioli J, Bresin E, Mossali C, Pianetti G, Gamba S, Daina E, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. 2010;5(10):1844-1859.
doi pubmed - Sallee M, Ismail K, Fakhouri F, Vacher-Coponat H, Moussi-Frances J, Fremaux-Bacchi V, Burtey S. Thrombocytopenia is not mandatory to diagnose haemolytic and uremic syndrome. BMC Nephrol. 2013;14:3.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.