

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website http://www.thejh.org |
Case Report
Volume 1, Number 1, February 2012, pages 23-27
Cerebrovascular Disease as Initial Clinical Presentation in a Patient With Idiopathic Thrombotic Thrombocytopenic Purpura: A Case Report
Diogenes Valderramaa, Jose Orsinia, b, Carlo Mainardia, Hilary Maa
aDepartment of Medicine, New York University School of Medicine at Woodhull Medical and Mental Health Center, 760 Broadway, Brooklyn, NY 11206, USA
bCorresponding author: Jose Orsin
Manuscript accepted for publication November 2, 2011
Short title: Cerebrovascular Disease
doi: https://doi.org/10.4021/jh102e
| Abstract | ▴Top |
There are very few hematological emergencies that they are usually excluded from the list of differential diagnosis; nonetheless, if untreated, they can lead to devastating results. Thrombotic thrombocytopenic purpura (TTP) is a thrombotic microangiopathy with characteristic von Willebrand factor (vWF)-rich microthrombi affecting the arterioles and capillaries of multiple organs. It represents both a diagnostic and management challenge to clinicians. Timely recognition and institution of appropriate therapy are of paramount importance in optimizing outcome. Nevertheless, TTP may have an unpredictable behavior once is diagnosed because of its variable frequency of relapses and exacerbations.
Keywords: TTP; Plasma exchange; Thrombotic microangiopathy
| Introduction | ▴Top |
There are very few hematological emergencies that they are usually excluded from the list of differential diagnosis; nonetheless, if untreated, they can lead to devastating results. Thrombotic thrombocytopenic purpura (TTP) is a thrombotic microangiopathy with characteristic von Willebrand factor (vWF)-rich microthrombi affecting the arterioles and capillaries of multiple organs. It represents both a diagnostic and management challenge to clinicians. Timely recognition and institution of appropriate therapy are of paramount importance in optimizing outcome. Nevertheless, TTP may have an unpredictable behavior once is diagnosed because of its variable frequency of relapses and exacerbations.
| Case Report | ▴Top |
A 48-year-old African American female presented to the emergency department complaining of right occipital headache and left upper extremity weakness and numbness for 4 days. She also referred left eye pain and slurred speech for 1 day. Her past medical history was remarkable for essential systemic hypertension, diabetes mellitus, and osteoarthritis. She underwent a non-complicated abdominal hysterectomy for uterine fibroids few years before admission. She claimed to be compliant with her home medications that include: amlodipine, benazepril, glimepiride, and occasional non-steroidal antiinflamatory drugs. She denied blurry vision, nausea, vomiting, fever, chills, or confusion. She completed a total of 7-day course of oral ciprofloxacin for a presumptive upper respiratory tract infection, after returning from her native Trinidad & Tobago six months prior to the presenting symptoms.
Her vital signs on admission were: blood pressure of 138/92 mmHg, heart rate of 112 beats/minute, respiratory rate of 19 breaths/minute, and a temperature of 98.4 °F.
Remarkable findings on physical examination include a decreased muscular strength (3/5) on the left upper extremity. There was no neck rigidity or tenderness to palpation over the temporal areas.
The complete blood count revealed a white blood cell count of 12,300/mm3 (4.8 - 10.8), a hemoglobin level of 7.2 g/dL (12.0 - 16.0), a hematocrit level of 19.2% (37 - 47), and a platelet count of 25,000/mm3 (130,000 - 400,000). Ferritin level was 477.9 µg/L (10 - 291), iron level was 53 µg/dL (60 - 180), total iron binding capacity was 209 µg/dL (250 - 450), and a reticulocyte count was 12.1% (0.5 - 1.5). Lactate dehydrogenase (LDH) level was 663 IU/L (80 - 200). Serum creatinine, electrolytes, erythrosedimentation rate, liver function, antinuclear antibodies (ANA), antiplatelets antibodies, haptoglobin level, and coagulation studies were within normal limits. Her CD4 cell count was 349/µL (544 - 1894). She was scheduled to be tested for human immunodeficiency virus (HIV) as outpatient. Peripheral blood smear demonstrated the presence of fragmented red blood cells (Fig. 1). ADAMTS-13 activity level was <1% (68 - 163).
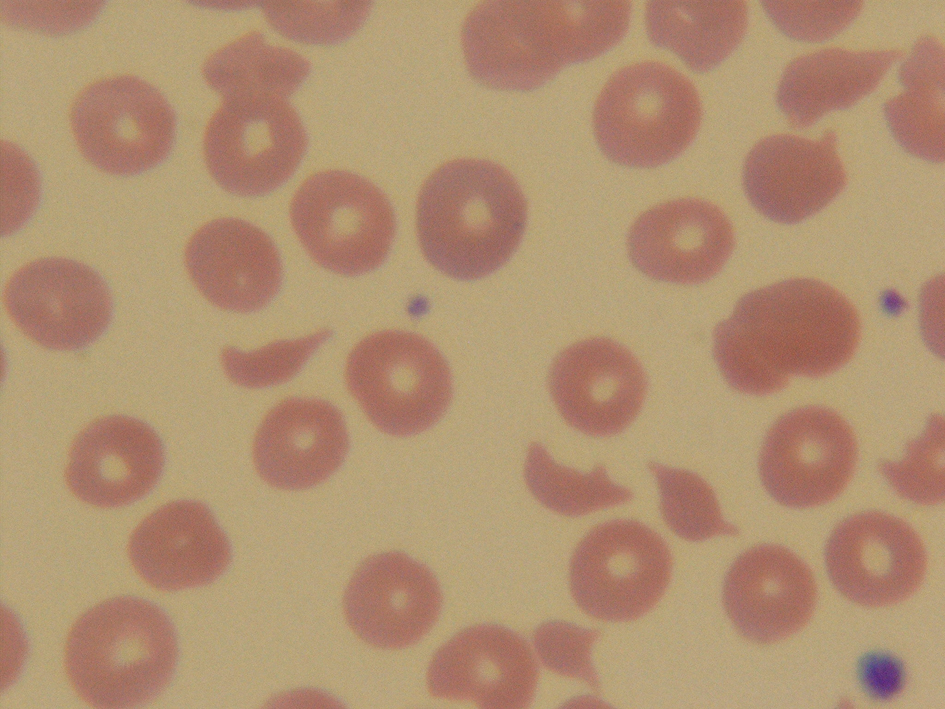 Click for large image | Figure 1. Peripheral blood smear demonstrated the presence of fragmented red blood cells. |
Chest radiography showed no infiltrates. Computed tomography of the head exhibited empty sella turcica without acute infarcts or hemorrhage.
She was admitted to a monitoring bed with the presumptive diagnosis of transient ischemic attack (TIA) or ischemic stroke. Plasma exchange therapy was initiated on day 1 for suspected thrombotic thrombocytopenic purpura (TTP). In addition, corticosteroids (prednisone 40 mg PO daily) were added to the therapy regimen. After the 2nd cycle of plasmapheresis, neurologic symptoms disappeared and deficits resolved, hemoglobin level and platelet count normalized, and lactate dehydrogenase level decreased. She completed a total of 3 cycles of plasmapheresis and was discharged home asymptomatic. She was followed in the Medicine clinic as outpatient after 3 days of discharge from the hospital. She was complaining of generalized weakness. Physical examination was remarkable for jaundice, but no neurologic deficits were found. She was readmitted to the hospital after laboratory results revealed a platelet count of 10,000/mm3, a total bilirrubin level of 4.1 mg/dl (0.0 - 1.5), a direct bilirrubin level of 3.3 mg/dl (0.0 - 0.5), and an LDH level of 1,642 IU/L. Coagulation studies were within normal limits. After 12 hours of admission, she developed cardiac arrest and expired.
Autopsy reported thrombotic thrombocytopenic purpura with congestive heart failure, bilateral pleural effusion, and fatty liver as main causes of death.
| Discussion | ▴Top |
Thrombotic thrombocytopenic purpura (TTP) is a life-threatening, severe multisystemic disorder of widespread microvascular thrombosis involving the capillaries and arterioles of different organs, characterized by fever, microangiopathic hemolytic anemia, and thrombocytopenia with or without neurological symptoms and impaired renal function [1, 2].
The pathophysiology of TTP is complex. At autopsy, widespread hyaline thrombi, accompanied by variable fibroblastic infiltration and endothelial overlay, are found in the terminal arterioles and capillaries of multiple organs, most extensively in the heart, brain, kidney, pancreas, spleen, mesentery, and adrenal glands. These thrombi are composed primarily of platelets and von Willebrand factor (vWF) [3, 4]. In addition to serving as the carrier of factor VIII, vWF mediates platelet adhesion and aggregation at sites of vessel injury. Under physiological conditions, the interaction between vWF and platelets is essential for the effective hemostasis at sites of arteriolar or capillary injury. When subjected to shear stress, endothelial vWF polymer is converted to plasma multimers by ADAMTS-13, a zinc metalloprotease that derives primarily from the hepatocytes [2, 5]. By cleaving vWF, ADAMTS-13 mitigates the tendency of vWF and platelets to form agglomerates in normal microcirculation. Thus, the physiological role of ADAMTS-13, the vWF cleaving protease, is to prevent intravascular platelet thrombosis [2]. Under the pathologic conditions of TTP, various intrinsic and extrinsic disorders precipitate ADAMTS-13 deficiency, including genetic mutations, autoimmune inhibitors or autoantibodies against ADAMTS-13, medications (ticlopidine, quinine, clopidogrel), HIV infection, sepsis, malignancy, disseminated intravascular coagulopathy (DIC), liver diseases, pregnancy, acute respiratory distress syndrome (ARDS), and Plasmodium falciparum infection [2, 6-10].
Many patients with TTP present with the triad of thrombocytopenia, microangiopathic hemolysis, and neurological abnormalities. Some of them may also have fever and renal abnormalities. However, it is important to note that neither the triad nor the pentad of presentation can be relied upon for the diagnosis of TTP. In practice, a constellation of thrombocytopenia and microangiopathic hemolysis should always raise the suspicion of TTP. Arterial hypertension and acute kidney injury requiring dialysis are uncommon [11]. It is increasingly recognized that patients with TTP may be asymptomatic, but with isolated thrombocytopenia requiring distinction from idiopathic thrombocytopenic purpura (ITP). It may also present with a transient ischemic attack (TIA) or stroke with or without hematological changes [2, 12]. TTP is a potential but infrequent cause of cerebrovascular disease in young adults. Usually, these patients present with headache, irrational behavior, seizures, and focal neurological deficits. Neurological abnormalities are multiple and secondary to microvascular platelet-fibrin thrombi that involve small arteries and capillaries [13]. In a series of 47 patients with acute TTP, the most common neuroradiologic finding was posterior reversible encephalopathy syndrome (PRES), while large ischemic infarctions and hemorrhage were uncommon [14]. During remission, neurocognitive deficits remain highly prevalent, are largely undetected by routine clinical evaluations, and may persist long after recovery from an acute episode of TTP [15, 16]. In the majority of patients with TTP, brain imaging studies do not evidence any abnormalities, as was the case with our patient. Because the diagnostic criteria of TTP are non-specific, the prompt recognition of the disease may be difficult. Differential diagnosis includes disseminated malignant neoplasias, systemic infections, malignant hypertension, systemic lupus erythematosus (SLE), and other thrombotic microangiopathies such as hemolytic-uremic syndrome (HUS) [17-19]. The clinical course of TTP is characterized by recurrent episodes in up to 50% of cases. Neurological symptoms are milder and its prevalence as well as the mortality is significantly lower in recurrences than in the first episode [20].
In general, thrombotic microangiopathies have been defined as having all the following alterations: microangiopathic hemolytic anemia (hemoglobin < 12 g/dL), negative Coombs test, undetectable serum haptoglobin levels (< 10 mg/dL), more than two fragmented red blood cells (schistocytes) in a 100 × microscopic field, increased LDH levels, thrombocytopenia (< 100,000/mm3), and organic dysfunction (renal and neurological) without signs of DIC [21, 22]. Based on laboratory findings, the differential diagnosis between TTP and other microangiopathies could be challenging. Immediate recognition of TTP is imperative because of the increased mortality, if not treated. Measuring ADAMTS-13 activity levels may not guarantee initial diagnosis and therapeutic decisions, but it is important for the prognosis. An ADAMTS-13 activity level < 5% - 10% seems to have increased specificity for TTP, but it does not identify all patients at risk for relapsing. ADAMTS-13 activity levels of > 10% makes the diagnosis of TTP unlikely in patients presenting with acute thrombocytopenia [2, 23, 24].
Therapeutic plasma exchange is the main therapy for patients with TTP [25]. Plasma exchange delivers elevated ADAMTS-13 dose without circulatory overload and removes antibodies to ADAMTS-13, recovering ADAMTS-13 activity. The treatment approach consists of a 1 to 1.5 plasma volume exchange with plasma daily until clinical symptoms have resolved and the platelet count has reached a normal level [24]. LDH levels should also be monitored since it reflects ongoing tissue ischemia as well as hemolysis [26]. Schistocytes measurements are not predictive of ability to maintain remission after cessation of therapy [27]. During remission of TTP, measurements of ADAMTS-13 levels may also be helpful. ADAMTS-13 activity levels < 5% - 10% are associated with high risk of relapse in the near future [28]. Although few schistocytes were found on the follow up peripheral blood smear, the patient discussed in this report demonstrated resolution of neurological deficits and normalization of the platelet count after the 2nd cycle of plasma exchange therapy. This patient did not have a follow up ADAMTS-13 activity level. In recurrences of TTP, platelet count is usually higher and the LDH levels lower than the first episode of the disease [20]. On readmission, our patient presented with more severe thrombocytopenia and higher LDH levels. There is evidence that the plasma exchange therapy would have only a transient effect on the presumably autoimmune basis of the disease, and that additional immunosuppressive therapy could lead to a longer-lasting response [29]. Treatment with immunosuppressive agents is reserved for patients suspected of having ADAMTS-13 autoimmune deficiency. Glucocorticoids are the immunosuppressive agents initially administered. Other agents such as rituximab and cyclosporine are used for more critically-ill patients and patients with recurrent disease [30, 31]. Aspirin is not used as adjuvant treatment, but is appropriate for patients with baseline cardiac or neurologic dysfunction and without severe thrombocytopenia. Other therapies used with variable success include cyclophosphamide, vincristine, azathioprine, intravenous immunoglobulins, and splenectomy. Despite the institution of appropriate treatment, mortality among patients with TTP remains approximately 15%. However, 50% of the deaths can be attributed to treatment-related complications [32]. Even after recognizing and treating the disease, patients have a significantly altered quality of life, with progressive memory loss and fatigue [33].
At the present time, the main clinical value of ADAMTS-13 activity assay is as a marker for outcome and the potential for relapse. Patients with severe deficiency of ADAMTS-13 experience higher rates of remission and lower mortality [34].
Rapid diagnosis and treatment are necessary for decreasing the risk of fatal outcome in patients with TTP. Clinicians should be familiar with the clinical presentation and laboratory abnormalities of TTP, in order to make early diagnosis and initiate appropriate therapy.
| References | ▴Top |
- George JN. How I treat patients with thrombotic thrombocytopenic purpura: 2010. Blood. 2010;116(20):4060-4069.
pubmed doi - Tsai HM. Pathophysiology of thrombotic thrombocytopenic purpura. Int J Hematol. 2010;91(1):1-19.
pubmed doi - Asada Y, Sumiyoshi A, Hayashi T, Suzumiya J, Kaketani K. Immunohistochemistry of vascular lesion in thrombotic thrombocytopenic purpura, with special reference to factor VIII related antigen. Thromb Res. 1985;38(5):469-479.
pubmed doi - Hosler GA, Cusumano AM, Hutchins GM. Thrombotic thrombocytopenic purpura and hemolytic uremic syndrome are distinct pathologic entities. A review of 56 autopsy cases. Arch Pathol Lab Med. 2003;127(7):834-839.
pubmed - Tsai HM, Sussman, II, Nagel RL. Shear stress enhances the proteolysis of von Willebrand factor in normal plasma. Blood. 1994;83(8):2171-2179.
pubmed - Sugio Y, Okamura T, Shimoda K, Matsumoto M, Yagi H, Ishizashi H, Niho Y, et al. Ticlopidine-Associated thrombotic thrombocytopenic purpura with an IgG-type inhibitor to von Willebrand factor-cleaving protease activity. Int J Hematol. 2001;74(3):347-351.
pubmed doi - Terrell DR, Williams LA, Vesely SK, Lammle B, Hovinga JA, George JN. The incidence of thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: all patients, idiopathic patients, and patients with severe ADAMTS-13 deficiency. J Thromb Haemost. 2005;3(7):1432-1436.
pubmed doi - Kremer Hovinga JA, Zeerleder S, Kessler P, Romani de Wit T, van Mourik JA, Hack CE, ten Cate H, et al. ADAMTS-13, von Willebrand factor and related parameters in severe sepsis and septic shock. J Thromb Haemost. 2007;5(11):2284-2290.
pubmed doi - Uemura M, Matsuyama T, Ishikawa M, Fujimoto M, Kojima H, Sakurai S, Ishii S, et al. Decreased activity of plasma ADAMTS13 may contribute to the development of liver disturbance and multiorgan failure in patients with alcoholic hepatitis. Alcohol Clin Exp Res. 2005;29(12 Suppl):264S-271S.
pubmed doi - Chang JC, Kathula SK. Various clinical manifestations in patients with thrombotic microangiopathy. J Investig Med. 2002;50(3):201-206.
pubmed doi - Tsai HM. The molecular biology of thrombotic microangiopathy. Kidney Int. 2006;70(1):16-23.
pubmed doi - Arboix A, Besses C. Cerebrovascular disease as the initial clinical presentation of haematological disorders. Eur Neurol. 1997;37(4):207-211.
pubmed doi - Stracciari A, Oiucci G, Bianchedi G, Beccari G. Computed tomographic evidence of cerebral focal ischemic lesion in thrombotic thrombocytopenic purpura. Arch Neurol. 1987;44(10):996.
pubmed doi - Burrus TM, Wijdicks EF, Rabinstein AA. Brain lesions are most often reversible in acute thrombotic thrombocytopenic purpura. Neurology. 2009;73(1):66-70.
pubmed doi - Cataland SR, Scully MA, Paskavitz J, Maruff P, Witkoff L, Jin M, Uva N, et al. Evidence of persistent neurologic injury following thrombotic thrombocytopenic purpura. Am J Hematol. 2011;86(1):87-89.
pubmed doi - Kennedy AS, Lewis QF, Scott JG, Kremer Hovinga JA, Lammle B, Terrell DR, Vesely SK, et al. Cognitive deficits after recovery from thrombotic thrombocytopenic purpura. Transfusion. 2009;49(6):1092-1101.
pubmed doi - Francis KK, Kalyanam N, Terrell DR, Vesely SK, George JN. Disseminated malignancy misdiagnosed as thrombotic thrombocytopenic purpura: A report of 10 patients and a systematic review of published cases. Oncologist. 2007;12(1):11-19.
pubmed doi - Robboy SJ, Salisbury K, Ragsdale B, Bobroff LM, Jacobson BM, Colman RW. Mechanism of Aspergillus-induced microangiopathic hemolytic anemia. Arch Intern Med. 1971;128(5):790-793.
pubmed doi - Egan JA, Bandarenko N, Hay SN, Paradowski L, Goldberg R, Nickeleit V, Brecher ME. Differentiating thrombotic microangiopathies induced by severe hypertension from anemia and thrombocytopenia seen in thrombotic thrombocytopenia purpura. J Clin Apher. 2004;19(3):125-129.
pubmed doi - Lotta LA, Mariani M, Consonni D, Mancini I, Palla R, Maino A, Vucelic D, et al. Different clinical severity of first episodes and recurrences of thrombotic thrombocytopenic purpura. Br J Haematol. 2010;151(5):488-494.
pubmed doi - George JN. Clinical practice. Thrombotic thrombocytopenic purpura. N Engl J Med. 2006;354(18):1927-1935.
pubmed doi - Ruutu T, Barosi G, Benjamin RJ, Clark RE, George JN, Gratwohl A, Holler E, et al. Diagnostic criteria for hematopoietic stem cell transplant-associated microangiopathy: results of a consensus process by an International Working Group. Haematologica. 2007;92(1):95-100.
pubmed doi - Polito MG, Kirsztajn GM. Thrombotic microangiopathies: thrombotic thrombocytopenic purpura / hemolytic uremic syndrome. J Bras Nefrol. 2010;32(3):303-315.
pubmed doi - Kiss JE. Thrombotic thrombocytopenic purpura: recognition and management. Int J Hematol. 2010;91(1):36-45.
pubmed doi - Rock GA, Shumak KH, Buskard NA, Blanchette VS, Kelton JG, Nair RC, Spasoff RA. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian Apheresis Study Group. N Engl J Med. 1991;325(6):393-397.
pubmed doi - Cohen JA, Brecher ME, Bandarenko N. Cellular source of serum lactate dehydrogenase elevation in patients with thrombotic thrombocytopenic purpura. J Clin Apher. 1998;13(1):16-19.
pubmed doi - Egan JA, Hay SN, Brecher ME. Frequency and significance of schistocytes in TTP/HUS patients at the discontinuation of plasma exchange therapy. J Clin Apher. 2004;19(4):165-167.
pubmed doi - Jin M, Casper TC, Cataland SR, Kennedy MS, Lin S, Li YJ, Wu HM. Relationship between ADAMTS13 activity in clinical remission and the risk of TTP relapse. Br J Haematol. 2008;141(5):651-658.
pubmed doi - Thorpe CM, Hurley BP, Lincicome LL, Jacewicz MS, Keusch GT, Acheson DW. Shiga toxins stimulate secretion of interleukin-8 from intestinal epithelial cells. Infect Immun. 1999;67(11):5985-5993.
pubmed - George JN, Woodson RD, Kiss JE, Kojouri K, Vesely SK. Rituximab therapy for thrombotic thrombocytopenic purpura: a proposed study of the Transfusion Medicine/Hemostasis Clinical Trials Network with a systematic review of rituximab therapy for immune-mediated disorders. J Clin Apher. 2006;21(1):49-56.
pubmed doi - Cataland SR, Jin M, Ferketich AK, Kennedy MS, Kraut EH, George JN, Wu HM. An evaluation of cyclosporin and corticosteroids individually as adjuncts to plasma exchange in the treatment of thrombotic thrombocytopenic purpura. Br J Haematol. 2007;136(1):146-149.
pubmed doi - George JN. The thrombotic thrombocytopenic purpura and hemolytic uremic syndromes: evaluation, management, and long-term outcomes experience of the Oklahoma TTP-HUS Registry, 1989-2007. Kidney Int Suppl. 2009;(112):S52-54.
pubmed - Lewis QF, Scott JG, Kremer Hovinga JA. Neurocognitive impairment following recovery from ADAMTS-13-deficient thrombotic thrombocytopenic purpura. Blood. 2007;110:395.
- Raife T, Atkinson B, Montgomery R, Vesely S, Friedman K. Severe deficiency of VWF-cleaving protease (ADAMTS13) activity defines a distinct population of thrombotic microangiopathy patients. Transfusion. 2004;44(2):146-150.
pubmed doi
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.