

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website http://www.thejh.org |
Case Report
Volume 5, Number 1, March 2016, pages 25-29
Fatal Case of Subcutaneous Panniculitis T-Cell Lymphoma Mimicking Polyarteritis Nodosa: A Case Report and Review of Literature
Rahul Sehgala, c, Ralph Yachouia, Jonathan Cutlanb
aDepartment of Rheumatology, Marshfield Clinic, Marshfield, WI, USA
bDepartment of Lab/Pathology, Marshfield Clinic, Marshfield, WI, USA
cCorresponding Author: Rahul Sehgal, Department of Rheumatology, Marshfield Clinic, 1000 North Oak Avenue, Marshfield, WI, USA
Manuscript accepted for publication January 21, 2016
Short title: Subcutaneous Panniculitis T-Cell Lymphoma
doi: http://dx.doi.org/10.14740/jh232w
| Abstract | ▴Top |
Subcutaneous panniculitis-like T-cell lymphoma (SPTL) is a rare subtype of cutaneous T-cell lymphoma. These cases can remain undiagnosed and are often treated with prolonged and ineffective immunosuppressive agents before a diagnosis of T-cell lymphoma is made. A 76-year-old white male presented with a 3-month history of fatigue, weight loss, night sweats, low grade fevers, gradual cognitive decline, and new onset skin lesions. Skin examination revealed mildly tender, indurated, dull red subcutaneous nodules on face, chest, back, upper, and lower extremities, most numerous on the chest and back. A punch biopsy from one of the skin nodules revealed small and medium vessel vasculitis with associated neutrophilic lobular panniculitis. A full thickness skin biopsy showed inflammatory infiltrate composed predominantly of atypical small to medium sized lymphocytes with hyperchromatic nuclei. A diagnosis of T-cell lymphoma was made on clinical presentation, histopathology, immunohistochemical and molecular studies. SPTL can mimic autoimmune diseases including primary vasculitis disorder in presentation. A superficial skin biopsy may be sufficient in diagnosing SPTL; however, presence of vasculitis on a superficial punch biopsy specimen should be followed by deep incisional biopsy (sometimes multiple) in all cases with atypical presentation or those who do not respond appropriately to treatment.
Keywords: Subcutaneous panniculitis T-cell lymphoma; Polyarteritis nodosa; Panniculitis; Bone marrow/pathology
| Introduction | ▴Top |
Subcutaneous panniculitis-like T-cell lymphoma (SPTL) of alpha/beta subtype is a distinct type of peripheral T-cell lymphoma involving the subcutaneous tissue. SPTL represents less than 1% of all primary cutaneous T-cell lymphomas. The skin manifestations of SPTL include subcutaneous nodules and plaques affecting the extremities and trunk, less commonly the face. These may be accompanied by B symptoms such as fever, weight loss and fatigue. Histopathologically, SPTL is characterized by lobular lymphocytic panniculitis. The differential diagnosis of SPTL is broad and includes benign and malignant lesions and autoimmune diseases such as primary systemic vasculitis. To our knowledge, we present the first description of SPTL masquerading as polyarteritis nodosa (PAN).
| Case Report | ▴Top |
A 76-year-old Caucasian male with a past medical history of hypertension and atrial fibrillation, on anticoagulation therapy with warfarin, presented to an emergency department with a 3-month history of fatigue, 40 pound weight loss, night sweats, low grade fevers, gradual cognitive decline, and new onset skin lesions. He denied any recent travel, sick contacts, infections, new medications, joint or muscle pains. He quit smoking over 10 years prior and denied illicit drug use. His family history was positive for leukemia in a brother and multiple myeloma in a sister. Vital signs on admission included blood pressure 119/77 mm Hg, regular heart rate at 68 beats/min, respiratory rate 20 breaths/min, and temperature 99.7 °F. On physical examination, the patient appeared malnourished and lethargic. Skin examination revealed mildly tender, indurated, dull red subcutaneous nodules on face, chest, back, upper, and lower extremities (Fig. 1). The lesions were most numerous on his chest and back with sparing of the palms, soles, and oral mucosa. Neurologic examination showed an ill-appearing male who was alert and oriented × 3. He had poor recollection at 5 min, but was able to speak fluently without any clear aphasia or neglect. Motor strength was 5/5 throughout, reflexes 2+ and light touch, and proprioception was symmetric in four extremities. The remainder of physical examination was unremarkable.
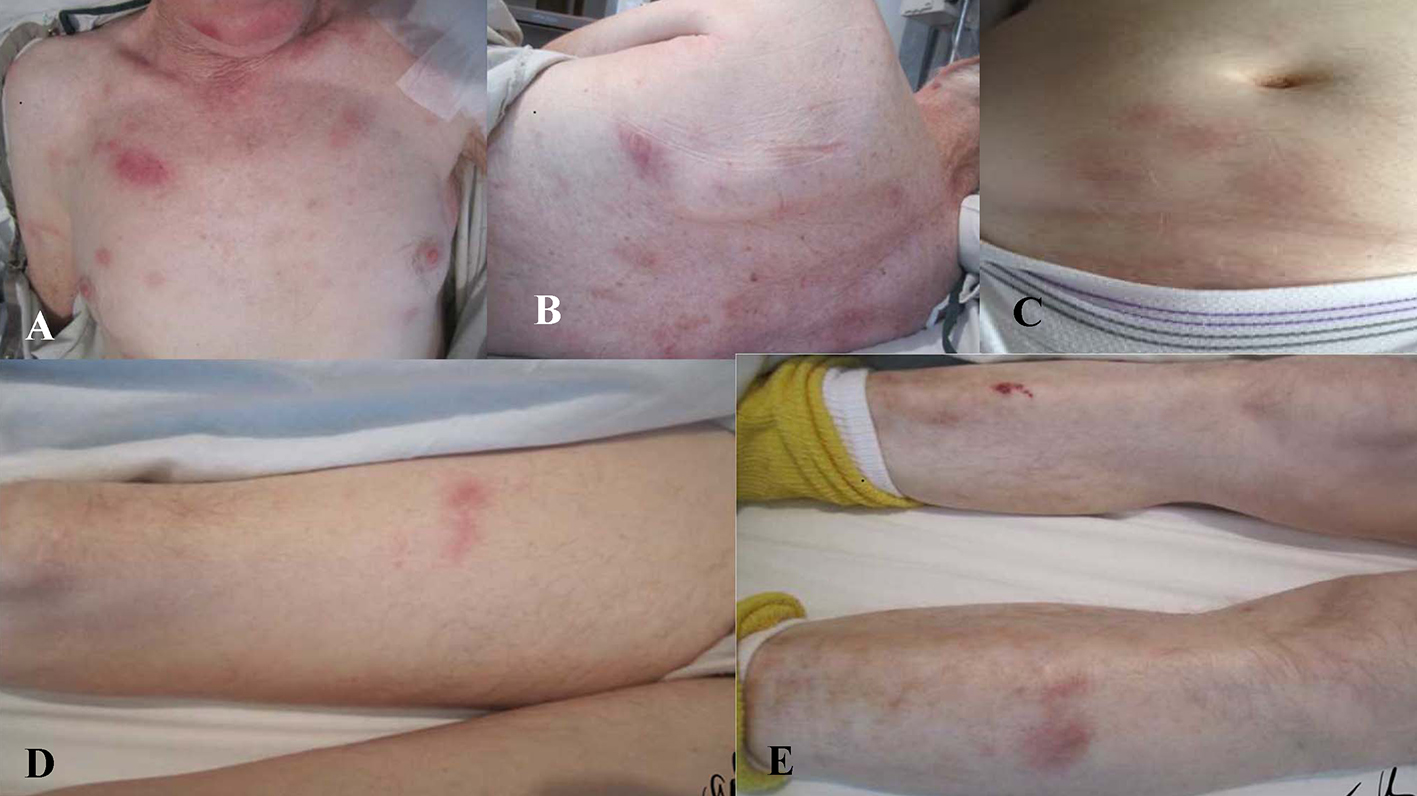 Click for large image | Figure 1. Mildly tender, indurated, dull red, subcutaneous nodules present on (A) chest, (B) trunk, (C) abdomen, and (D, E) extremities. |
Routine laboratory workup showed white blood cell count of 2,300/μL (nl: 4.8 - 10.8), hemoglobin 11.0 g/dL (nl: 14 - 16), platelet count 147,000/μL (nl: 130,000 - 400,000), albumin 2.5 g/dL (nl: 3.4 - 5), lactate dehydrogenase 393 U/L (nl: 81 - 190), serum ferritin 2,229 ng/mL (nl: 26 - 388), haptoglobin 13 mg/dL (nl: 41 - 230), antinuclear antibody test (ANA) 80 speckled pattern (nl < 80), and total alkaline phosphatase 90 IU/L (nl: 45 - 115); serologies including anti-double-stranded DNA antibody, rheumatoid factor, antineutrophil cytoplasmic antibody (ANCA), angiotensin converting enzyme (ACE), alpha 1 antitrypsin level and serum cryoglobulins were negative; complement levels were normal. Cerebral spinal fluid (CSF) analysis was normal with absent oligoclonal bands. Infectious disease workup including testing for latent tuberculosis (Q-gold), cultures of the blood, urine, and CSF, hepatitis B and C serologies, HIV infection, fungal testing for histoplasma and blastomyces antigen in urine were all negative. Kappa/lambda free light chain ratio was 0.65 (nl: 0.26 - 1.65). Quantitative immunoglobulins assay showed normal levels of immunoglobulins G, A, and M. Urine immunofixation showed presence of kappa Bence-Jones proteins. Urine protein/creatinine ratio was normal.
A computerized tomography (CT) scan of the chest, abdomen, and pelvis did not reveal any evidence for malignancy or hilar adenopathy. Magnetic resonance imaging of the brain did not reveal any discrete mass lesions/hemorrhage/acute ischemia, or abnormal enhancement. Bone marrow biopsy showed trilineage hematopoietic maturation and no convincing evidence for involvement by a neoplastic process. The patient did not meet criteria for hemophagocytic syndrome. A superficial punch biopsy from one of the skin nodules revealed small and medium vessel vasculitis with associated neutrophilic lobular panniculitis (Fig. 2). Cutaneous PAN was favored as a diagnosis.
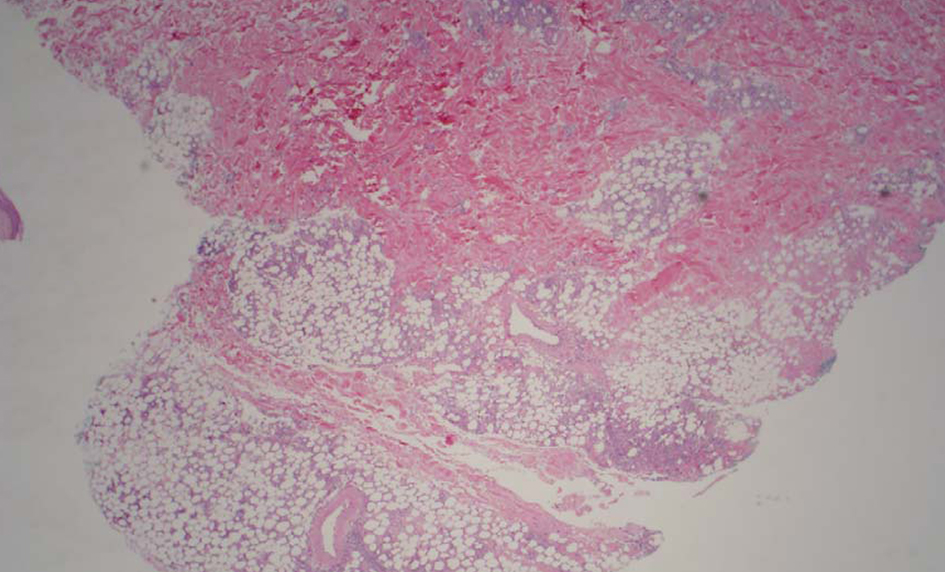 Click for large image | Figure 2. Low power image shows moderate subcutaneous inflammation localized to the lobules (lobular panniculitis). |
A full thickness skin biopsy was subsequently performed and showed inflammatory infiltrate composed predominantly of atypical small to medium sized lymphocytes with hyperchromatic nuclei, which in areas rimmed adipocytes. There was an abundance of nuclear debris with associated macrophages. Given the atypical lymphoid infiltrate, additional immunohistochemical studies were performed. The lymphoid infiltrate appeared to be predominantly CD3 positive T cells that were CD8 positive (Fig. 3A, B). CD4 also labeled some histiocytes and background T cells, but the CD8 preferentially labeled the atypical lymphocytes. The atypical lymphocytes failed to label with CD56. CD20 failed to label the lymphocytes. In addition, a T-cell receptor gene rearrangement showed the lymphocytes were clonal. A T-cell receptor (TCR)-BF1 immunohistochemical study confirmed an alpha-beta rather than gamma-delta T-cell origin. Molecular confirmation of T-cell lymphoma was made on T-cell receptor gene rearrangement studies. These findings favor an alpha-beta T cell, rather than a gamma-delta T cell (Fig. 3C).
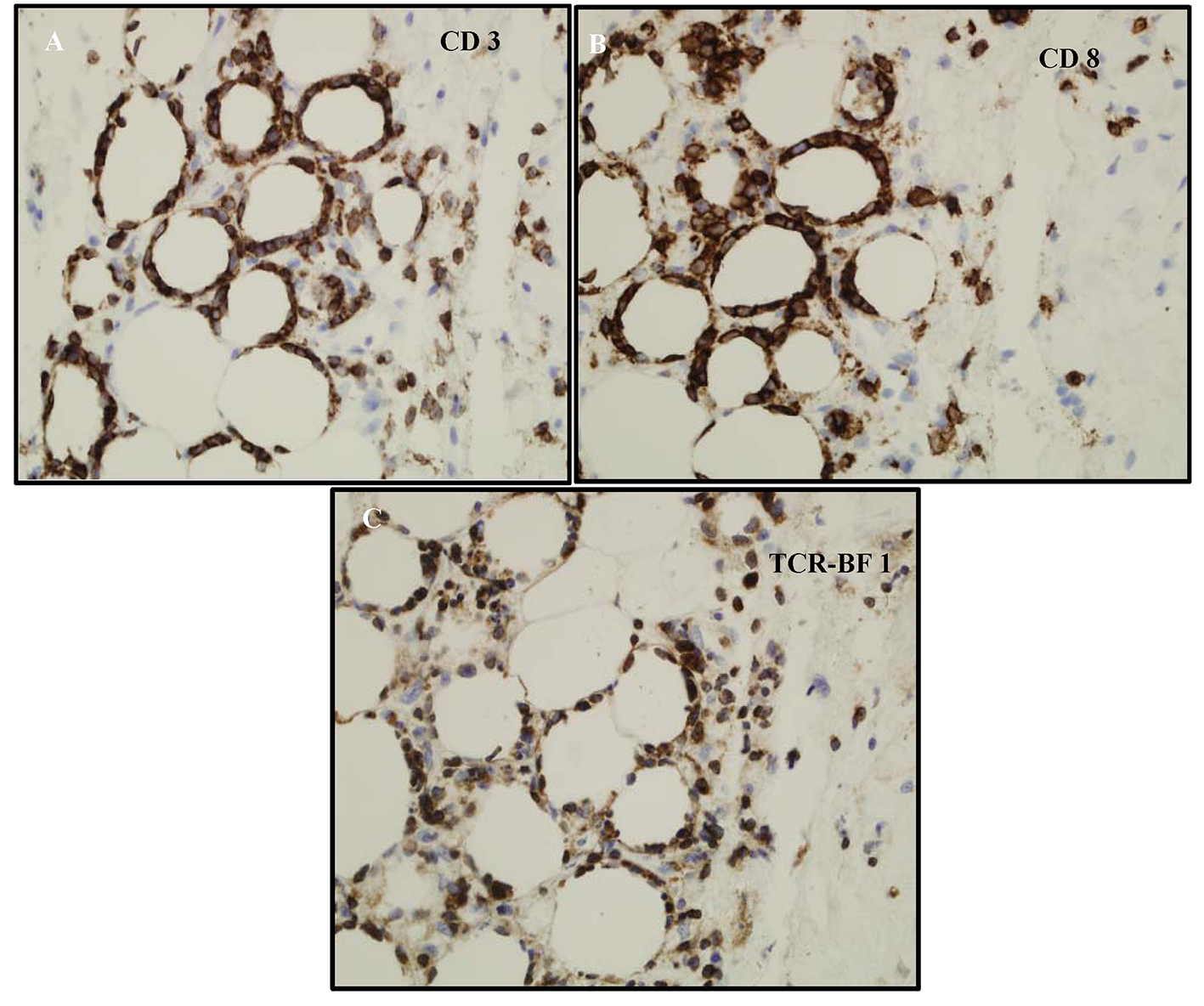 Click for large image | Figure 3. (A) CD3 labels the lymphocytes-supporting T cells. (B) Phenotype of the cells ends up being CD3+, CD8+, CD4-, CD56-, and TCR-BF1 positive. It appears to be mature T-cell phenotype with CD8 and derived from alpha/beta T-cells not gamma/delta. (C) The atypical lymphocytes appear to label with TCR-BF1 immunohistochemical study in keeping with an alpha beta T-cell, rather than a gamma delta T-cell origin. |
Fludeoxyglucose18 (FDG) positron emission tomography (PET)/CT scan was consistent with innumerable, very widespread areas of lymphoma, predominating within the superficial soft tissues of the entire visualized portion of the body and also involving deeper structures/organs, including the right temporalis muscle, the mediastinum, the stomach, the parapancreatic retroperitoneum, both adrenal glands, left perirenal/left iliopsoas fat, and the perirectal fat.
A bone marrow biopsy revealed trilineage hematopoietic maturation but no convincing evidence for a neoplastic process. The patient’s skin lesions improved on chemotherapy with cyclophosphamide, adriamycin, vincristine, and prednisone (CHOP) regimen and his pancytopenia resolved. However, he continued to have a gradual decline in his mental status, disease progression, infectious complications, and failure to thrive. He died approximately 3 months from the time of SPTL diagnosis.
| Discussion | ▴Top |
SPTL belongs to a group of primary cutaneous T-cell lymphoma characterized by subcutaneous nodules and plaques affecting the extremities and face and may be accompanied by fever, weight loss and fatigue [1]. Clinically, the lesions resemble those of panniculitis, usually affecting the trunk and extremities and sparing the face. As per current diagnostic criteria, SPTL is alpha/beta (CD4-/CD8+ and CD56-) positive based on the cell surface receptors (TCR) carried by neoplastic cells. The gamma/delta (CD4-, CD8-, and CD56+) type is now classified as a separate entity called primary cutaneous gamma delta T-cell lymphoma (TCL). Bone marrow involvement can occur with either phenotype of SPTL, though the majority of cases are confined to subcutaneous adipose tissue. A variety of diseases, especially dermatological and rheumatic conditions, may have an appearance and presentation similar to SPTL. These include conditions such as septal panniculitis (erythema nodosum), lobular panniculitides (erythema induratum, nodular vasculitis, and lupus panniculitis), lobular and septal panniculitis (alpha-1 antitrypsin deficiency panniculitis), infectious panniculitis, pyoderma gangrenosum, systemic vasculitis and inflammatory myopathies [2-5].
Histopathologic examination shows subcutaneous infiltrates containing a mixture of neoplastic pleomorphic cells of varying sizes and macrophages and rimming of individual fat cells by neoplastic cells are features of SPTL (Fig. 4). Immunophenotyping and clonal TCR gene rearrangement are other helpful tests in establishing a diagnosis. The constellation of above findings and demonstration of rimming of adipocytes by atypical lymphocytes with hyperchromatic nuclei establishes a diagnosis of SPTL. Prognosis of SPTL seems more favorable compared to TCL and depends upon hemophagocytosis in bone marrow (5-year survival of 42% vs. 82% with or without hemophagocytic syndrome (HS) in SPTL and 11% in TCL) [6]. Therapy for SPTL has not been well defined, although immunosuppressive agents like prednisone or multiagent chemotherapy have all been used for treatment.
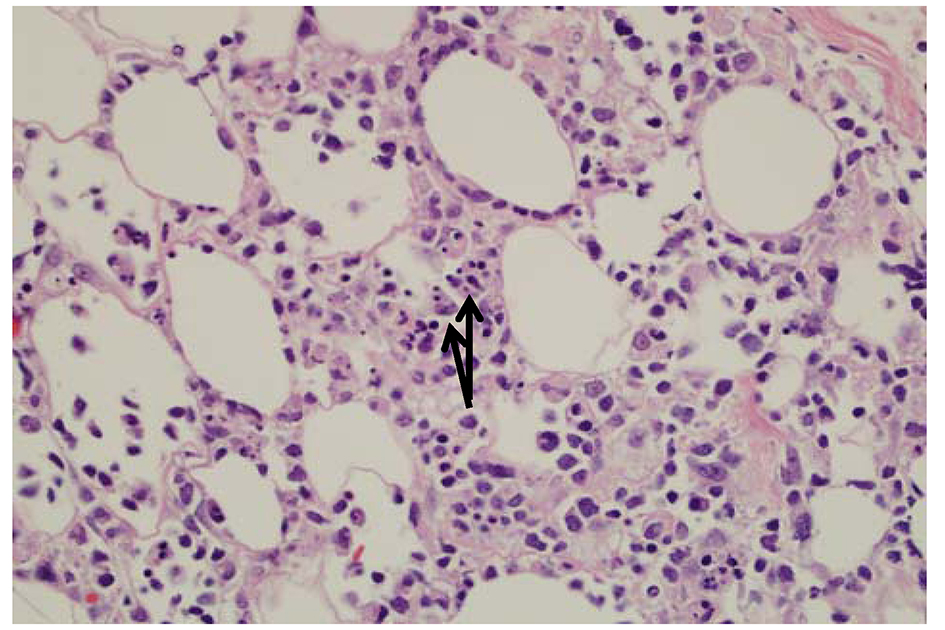 Click for large image | Figure 4. The fat is involved by an atypical lymphoid infiltrate with rimming of atypical lymphocytes around adipocytes. There are occasional histiocytes that engulf the lymphocytes, so called “bean bag” cells by arrows. |
The presence of similar cutaneous manifestations, constitutional symptoms, and absence of characteristic autoantibody makes diagnosis of PAN, a challenge. The diagnosis can be easily missed if punch biopsy alone is utilized for evaluating the skin lesions, as it only includes epidermis and superficial dermis. The presence of perivascular inflammatory infiltrate with features of vascular destruction (vasculitis) can be non-specific and be secondary to necrotizing panniculitis involving the adjacent vessel. This may give a false appearance of a primary vasculitic disorder (Fig. 5A, B, C). Evaluation of an area away from extensive necrosis reveals dense infiltrate of atypical lymphocytes and absence of vasculitis (Fig. 5D). A full thickness skin biopsy helps in diagnosing and distinguishing SPTL from other rheumatic diseases including vasculitis, myopathies, and lupus panniculitis. A deep incisional biopsy including subcutaneous fat also allows histologic analysis of an entire lesion and performance of multiple studies from a single biopsy site. Histologic evidence of inflammation of a medium- or small-sized artery by a mixed infiltrate of neutrophils, lymphocytes, and macrophages is usually seen in PAN. SPTL, on the other hand, is associated with rimming of fat cells by neoplastic lymphocytes. An astute histopathologist can make the distinction based on histopathology, immunophenotyping, and clonal TCR gene rearrangement. Bone marrow involvement in SPTL is rare [7-9]. Features such as splenomegaly, pancytopenia, elevated lactate dehydrogenase, ferritin, and triglycerides are manifested in HS, while PAN is usually accompanied by anemia, thrombocytosis, and elevated acute phase reactants (erythrocyte sedimentation rate and C-reactive protein). There are only four cases of PAN with HS described in literature [10-12]. In two of those cases, infection triggered the development of HS. In one case, both infection and onset of systemic disease were thought to play the role, while in another, HS developed after several years of undergoing successful treatment for PAN. Skin manifestations in PAN include livedo reticularis, nodules, and ulcerations that can be very painful, primarily involving the lower extremities, and rarely leading to frank ischemia of digits. This is in contrast to the multiple, painless (very rarely painful), erythematous indurated plaques and nodules seen in SPTL, which do not ulcerate. These can involve the upper and lower extremities as well as the trunk, face, and neck.
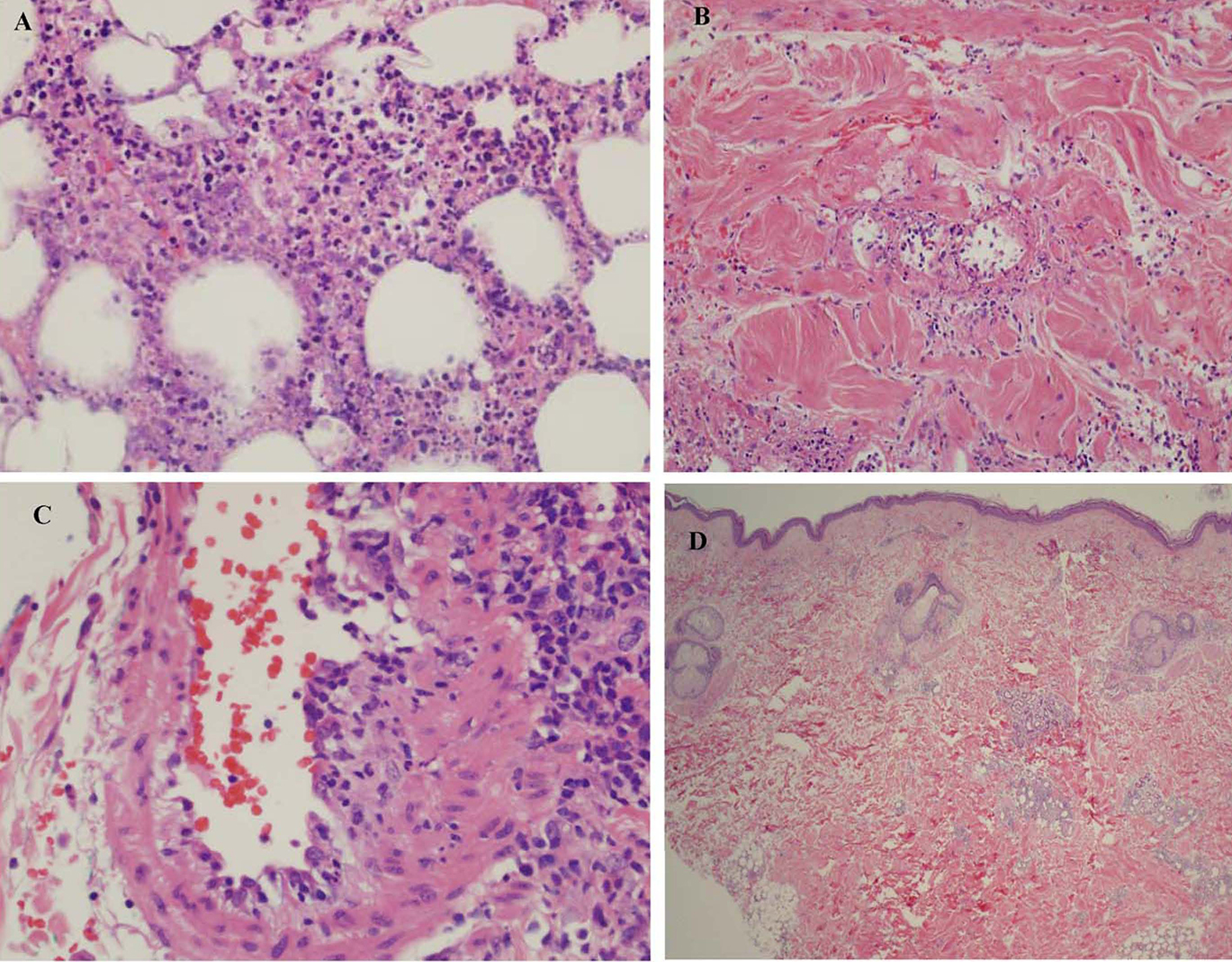 Click for large image | Figure 5. (A) Subcutaneous fat showing extensive necrotizing panniculitis with predominantly neutrophilic inflammation. (B) There is evidence of vasculitis within the subcutaneous septa. (C) A medium sized blood vessel shows some neutrophilic inflammation. (D) Biopsy shows less inflammation involving the subcutis and no extensive necrosis or vasculitis. |
In summary, SPTL can mimic various rheumatologic diseases. In a series of 11 cases, an autoimmune disease was the initial diagnosis, but finally discovered to be SPTL [5]. To the best of our knowledge, we present the first description of SPTL masquerading as PAN with a very aggressive and fatal course. SPTL can remain undiagnosed and often treated with prolonged and ineffective immunosuppressive agents. The presence of vasculitis on a superficial punch biopsy specimen must be followed by deep incisional biopsy (sometimes multiple) in all cases with atypical presentation or those who do not respond appropriately to systemic corticosteroids.
Financial Disclosures
None.
| References | ▴Top |
- Gonzalez CL, Medeiros LJ, Braziel RM, Jaffe ES. T-cell lymphoma involving subcutaneous tissue. A clinicopathologic entity commonly associated with hemophagocytic syndrome. Am J Surg Pathol. 1991;15(1):17-27.
doi pubmed - Chiu HY, He GY, Chen JS, Hsiao PF, Hsiao CH, Tsai TF. Subcutaneous panniculitis-like T-cell lymphoma presenting with clinicopathologic features of dermatomyositis. J Am Acad Dermatol. 2011;64(6):e121-123.
doi pubmed - Arps DP, Patel RM. Lupus profundus (panniculitis): a potential mimic of subcutaneous panniculitis-like T-cell lymphoma. Arch Pathol Lab Med. 2013;137(9):1211-1215.
doi pubmed - Kao GF, Resh B, McMahon C, Gojo I, Sun CC, Phillips D, Zhao XF. Fatal subcutaneous panniculitis-like T-cell lymphoma gamma/delta subtype (cutaneous gamma/delta T-cell lymphoma): report of a case and review of the literature. Am J Dermatopathol. 2008;30(6):593-599.
doi pubmed - Yi L, Qun S, Wenjie Z, Wen Z, Jian L, Yan Z, Fengchun Z. The presenting manifestations of subcutaneous panniculitis-like T-cell lymphoma and T-cell lymphoma and cutaneous gammadelta T-cell lymphoma may mimic those of rheumatic diseases: a report of 11 cases. Clin Rheumatol. 2013;32(8):1169-1175.
doi pubmed - LeBoit PE, Burg G, Weedon D, Sarasin A, eds. World Health Organization Classification of Tumours: Pathology and Genetics Skin Tumours. Lyon, France: IARC Press; 2006.
- Willemze R, Jansen PM, Cerroni L, Berti E, Santucci M, Assaf C, Canninga-van Dijk MR, et al. Subcutaneous panniculitis-like T-cell lymphoma: definition, classification, and prognostic factors: an EORTC Cutaneous Lymphoma Group Study of 83 cases. Blood. 2008;111(2):838-845.
doi pubmed - Ghobrial IM, Weenig RH, Pittlekow MR, Qu G, Kurtin PJ, Ristow K, Ansell SM. Clinical outcome of patients with subcutaneous panniculitis-like T-cell lymphoma. Leuk Lymphoma. 2005;46(5):703-708.
doi pubmed - Gao J, Gauerke SJ, Martinez-Escala ME, Guitart J, Nelson BP, Chadburn A, Peterson LC. Bone marrow involvement by subcutaneous panniculitis-like T-cell lymphoma: a report of three cases. Mod Pathol. 2014;27(6):800-807.
doi pubmed - Dhote R, Simon J, Papo T, Detournay B, Sailler L, Andre MH, Dupond JL, et al. Reactive hemophagocytic syndrome in adult systemic disease: report of twenty-six cases and literature review. Arthritis Rheum. 2003;49(5):633-639.
doi pubmed - Hayakawa I, Shirasaki F, Ikeda H, Oishi N, Hasegawa M, Sato S, Takehara K. Reactive hemophagocytic syndrome in a patient with polyarteritis nodosa associated with Epstein-Barr virus reactivation. Rheumatol Int. 2006;26(6):573-576.
doi pubmed - Harrold LR, Liu NY. Polyarteritis nodosa presenting as pancytopenia: case report and review of the literature. Rheumatol Int. 2008;28(10):1049-1051.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.