

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website http://www.thejh.org |
Case Report
Volume 5, Number 1, March 2016, pages 38-40
A Case of Hyperhemolytic Anemia
Alexandra Sokolovaa, c, Kamran Darabib
aNassau University Medical Center, East Meadow, NY, USA
bRidgeview Medical Center, Waconia, MN, USA
cCorresponding Author: Alexandra Sokolova, Internal Medicine Department, Nassau University Medical Center, 2201 Hempstead tpk, East Meadow, NY 11554, USA
Manuscript accepted for publication March 07, 2016
Short title: Hyperhemolytic Anemia
doi: http://dx.doi.org/10.14740/jh266e
| Abstract | ▴Top |
Delayed hemolytic transfusion reaction (DHTR) is a well-known complication of red blood transfusion. Occasionally, native red cell hemolysis can complicate transfused red cell hemolysis, resulting in a lower post-transfusion hemoglobin. This condition is known as hyperhemolysis syndrome, and is more common after a blood transfusion among patients with sickle cell disease and thalasemia. The occurrence of this syndrome in patients without underlying hemoglobinopathies is rare. A 74-year-old female with a previous history of colonic adenocarcinoma presented to a local hospital with a complaint of diarrhea. She was found to be anemic with hemoglobin of 8.4 g/dL. Her blood was group A, Rh-negative and she received two units of compatible packed red blood cells, after a negative antibody screen was documented. She returned to the hospital 12 days later with complaints of lethargy and fatigue. She was found to be anemic with hemoglobin of 7.8 g/dL. Antibody screening was positive for anti-K antibody. Hemolysis persisted despite receiving K-negative red blood cells, twice during the next 30 days. Both times, her hemoglobin decreased to a level lower than the pre-transfusion value. No other source of bleeding could be identified and a normal hemoglobin electrophoresis was noted. She responded well to 4 weeks of oral prednisone, with no further hemolysis after discontinuation of prednisone therapy. Hyperhemolysis syndrome can occur in patients without hemoglobinopathy, and oral prednisone is an effective treatment.
Keywords: Delayed serologic transfusion reaction; Delayed hemolytic transfusion reaction; Red blood cells; Hemoglobin; Hematocrit; Direct antiglobulin test
| Introduction | ▴Top |
Delayed serologic transfusion reaction (DSTR) is defined as sensitization against red blood cells without evidence of active hemolysis. Delayed hemolytic transfusion reaction (DHTR) is defined as DSTR with clinical or biochemical evidence of hemolysis. Pineda et al reviewed 201,445 allogenic red blood cell (RBC) units transfused and identified DSTR with an incidence of 1/1,612 and DHTR with incidence of 1/6,715 [1]. Occasionally, hemolysis of not only the transfused cells but also of patient’s native red blood cells has been reported. This condition, termed hyperhemolysis syndrome, is well reported in patients with sickle cell disease [2] and other hemoglobinopathies including thalassemia [3]. Recent case reports of hyperhemolysis occurring in patients with anemia of chronic disease [4] and myelofibrosis [5] suggest that hyperhemolysis can occur in patients without hemoglobinopathies. We report a similar case of hyperhemolysis occurring in a patient with no underlying hemoglobinopathy.
| Case Report | ▴Top |
The patient is a 74-year-old female with blood group A, Rh-negative, who was transferred to our hospital from a small community hospital, due to the unavailability of cross match compatible blood. She was diagnosed with adenocarcinoma of the distal colon, for which she underwent low anterior resection of the sigmoid colon with primary anastamosis 6 months prior to presenting to our facility. Final staging was T3, N1, M0 for which she underwent eight cycles of 5-flourouracil, oxaliplatin and leucovorin chemotherapy. She also underwent adjuvant radiation therapy.
Prior to the diagnosis of colon cancer, the patient did not have any major medical problems. She had undergone cholecystectomy and hysterectomy (for benign reasons) in the remote past. During chemotherapy, she developed deep venous thrombosis of lower extremities for which coumadin therapy was started. She also developed a right-sided recurrent pleural effusion which required three thoracenteses for drainage. During chemotherapy, the patient also developed anemia, thought to be secondary to marrow suppression from chemotherapy treatment and required six units of packed red blood cells (PRBC) over a period of 5 months. The antibody screen preceding each transfusion was negative.
Three weeks prior to presenting to our facility, the patient was admitted to a local community hospital with complaint of diarrhea thought to be secondary to radiation enteritis. Incidentally, a moderate sized bilateral pleural effusion was identified. The patient underwent thoracentesis followed by chemical pleurodesis, and required two units of PRBC transfusion during her hospital stay. During the same admission, the patient underwent an upper and lower gastrointestinal endoscopy for anemia which revealed no bleeding source. On admission, her hemoglobin (Hb) was 8.3 g/dL (reference range: 12.0 - 16.0 g/dL) with a hematocrit (HCT) of 24.1% (reference range: 35.0-45.0%), and 6 days after transfusion her Hb was 12.1 g/dL (Fig. 1). Antibody screen prior to the transfusion was negative. Her INR was within therapeutic range.
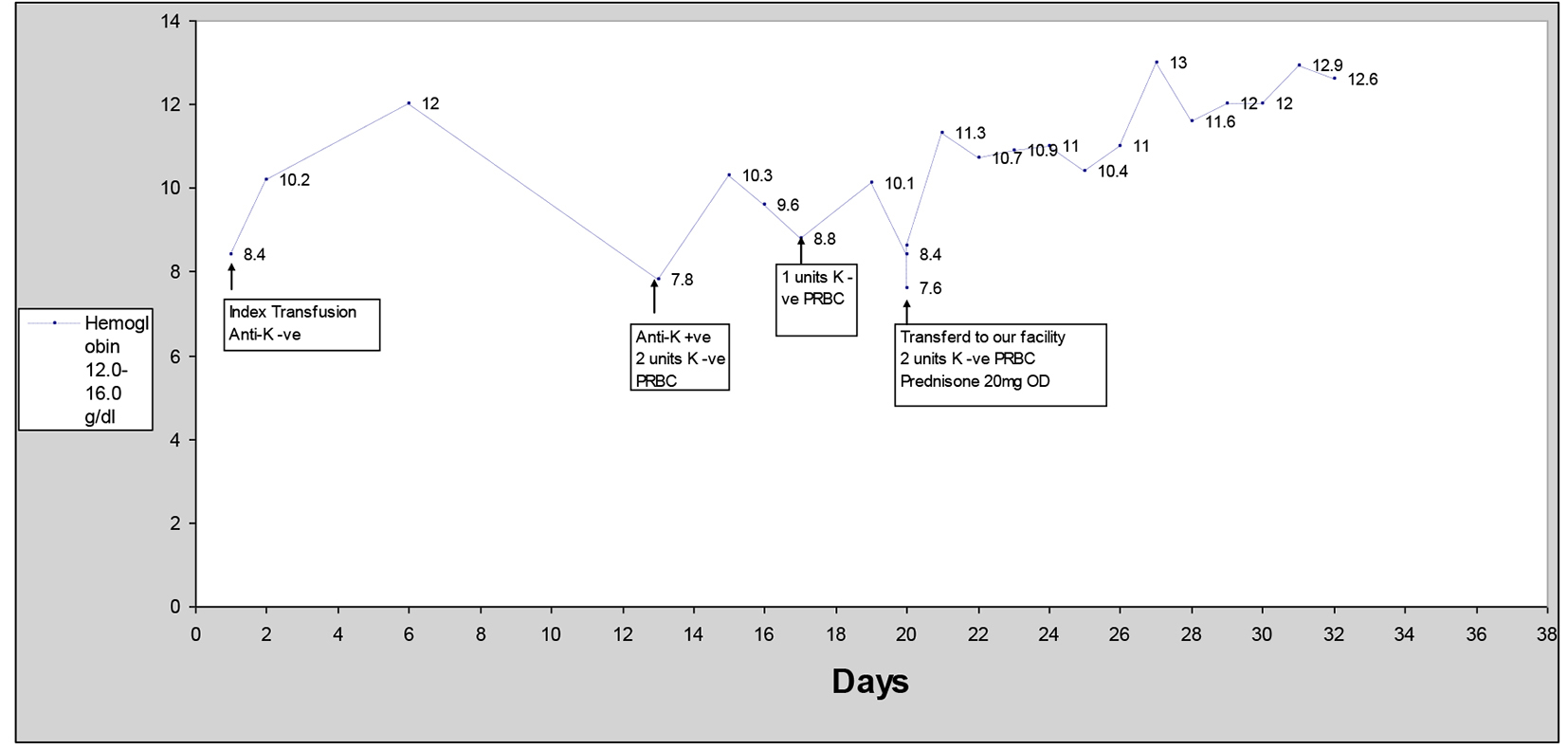 Click for large image | Figure 1. Hemoglobin vs. days after first PRBC transfusion. |
The patient was re-admitted to the same facility on day 12 after the last transfusion with profound weakness. Laboratory data showed anemia with Hb of 7.8 g/dL and HCT of 21.3%. No obvious source of a bleed could be identified. Total bilirubin was 1.45 mg/dL (reference range: 0.2 - 1.3 mg/dL) and ALT (reference range: 0 - 55 U/L) and AST (reference range: 0 - 37 U/L) were normal. Pre-transfusion antibody screen was positive for anti-K antibody. She received two units of K antigen-negative PRBC and was transferred to a swing bed facility for physical rehabilitation. Hb improved from 7.8 to 10.3 g/dL after the transfusion of two units of K-negative PRBC.
On day 17, her Hb decreased to 8.8 and two units of K-negative PRBC were transfused again which improved her Hb to 10.1 g/dL. On day 20, her Hb decreased again to 7.6 g/dL. Stool occult testing was again negative and given no obvious source of bleeding, coumadin was continued. She was transferred to our facility on day 20 due to unavailability of cross match compatible blood.
On transfer to our facility, the patient underwent repeat pre-transfusion screening with direct antiglobulin test (DAT/direct Coomb’s test) which was positive with anti-complement only (negative with anti-IgG), suggestive of IgM-mediated complement activation on the patient’s own RBCs. Antibody screen (indirect antiglobulin test) revealed that three out of three K-positive group O screening RBCs were 2+ reactive with IgG on day 20, suggesting the presence of anti-K IgG in the patient’s serum. Bilirubin on admission was 0.64 (Fig. 2). Moderate amount of spherocytes on peripheral smear was identified. Two units of K-negative PRBC were transfused which corrected her Hb to 11.3 g/dL. Other laboratory findings included elevated LDH of 240 U/L (reference range: 100 - 250 U/L) with reticulocyte count of 5.5% (reference range: 0.7-3.3%) and haptoglobin of 25 mg/dL (reference range: 30 - 200 mg/dL). She was started on prednisone 20 mg daily and no further transfusions were required.
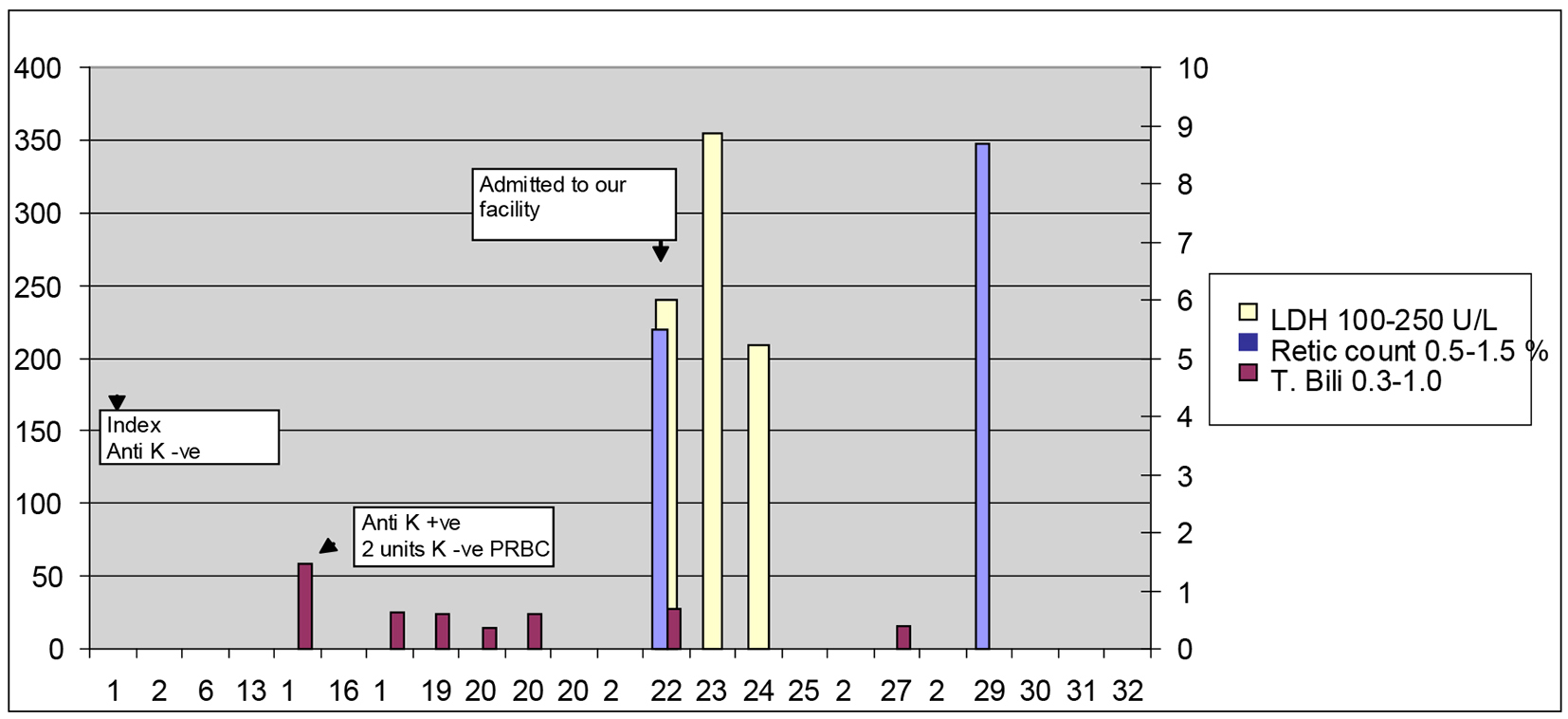 Click for large image | Figure 2. Total bilirubin, LDH and reticulocyte count vs. days after each PRBC transfusion. |
The patient improved over the next few days and her hemolytic indices normalized over time. Reticulocyte count reached highest on day 27 to 8.7%. Repeat reticulocyte count on day 59 was 1.3%. Haptoglobin on day 59 was 285 mg/dL. LDH continued to be elevated to 273 U/L on day 59 and decreased further to 231 U/L on day 112. Hb continued to be normal during this time period. Repeat DAT on day 59 was negative and continued to be negative on day 112. She only received 4 weeks of steroids which were slowly tapered over a period of 4 weeks. The patient also underwent Hb electrophoresis which was normal with no underlying hemoglobinopathy.
In summary, the patient’s Hb continued to drop despite K-negative blood transfusion. If K incompatibility would have been the only cause of hemolysis, then her Hb should have remained stable at that time. The fact that the patient continued to hemolyze RBCs even with K-negative blood transfusion raised the possibility of hyperhemolytic syndrome, when not only transfused but native RBCs are attacked by the host’s immune system. On multiple occasions, her Hb after transfusion was lower than pre-transfusion.
The patient did not require transfusions on prednisone therapy, and her Hb improved, this suggests that the production of the offending anti-K antibody by the immune system was effectively shut down, and no further hemolysis of the patient’s native RBCs occurred without further PRBC transfusion needed.
We postulate that the patient’s own RBCs were being hemolyzed as a result of bystander complement activation on the surface of the patient’s native RBCs, which was mediated by the anti-K antibodies in her serum.
| Discussion | ▴Top |
DHTRs are a frequent complication of transfusion therapy. Hyperhemolysis syndrome or bystander hemolysis is a poorly understood complication of transfusion therapy. This syndrome is characterized by post-transfusion Hb less than pre-transfusion Hb, signifying that not only the transfused cells but also patient’s own red cells hemolyzed during the process. The patient described here developed DHTR after receiving two units of PRBC. Twice during the 20-day course of illness, post-transfusion Hb decreased below pre-transfusion Hb. On day 20, DAT was positive with anti-complement but negative with anti-IgG while the indirect antiglobulin test detected a high titer anti-K alloantobody in the patient’s serum. We postulate that complement activation on the patient’s own RBCs occurred as a result of the production of crossreactive IgM antibodies while anti-K IgG was also present in the patient’s serum. In essence, the laboratory findings suggested that the transfused K-positive PRBCs had already been completely hemolyzed and crossreactive IgM antibodies were continuing to hemolyze the patient’s own RBCs despite transfusion with K-negative PRBCs essentially acting as an IgM autoantibody. Steroid therapy proved to be effective and we were able to discontinue steroids over a few weeks with no further evidence of hemolysis over several years of follow-up.
Previously, the occurrence of DHTR with hyperhemolysis has been documented in patients with hemoglobinopathies including sickle cell disease [2] and thalasemia [3]. Very few cases have been reported that have described the occurrence of this complication in patients with no known hemoglobinopathy including a patient with anemia of chronic disease [4] and another patient with myelofibrosis [5]. Our patient underwent Hb electrophoresis and no Hb abnormality was identified underlining the fact that hyperhemolysis can occur in patients without hemoglobinopathy.
The pathophysiology of this phenomenon is poorly understood. One proposed mechanism is the development of autoantibodies resulting in transient autoimmune hemolytic anemia. Another proposed mechanism previously proposed has been complement sensitization which leads to autologous RBC hemolysis in addition to transfused red cells [6].
Previously, steroid therapy and IVIG therapy have been used as treatment for hyperhemolysis [7]. Our patient responded well to 4 weeks of steroid therapy. We maintained close follow-up over 6 months during which her Hb remained normal.
Conclusion
Hyperhemolysis can be a life-threatening complication of transfusion therapy. It can occur in patients with no underlying hemoglobinopathy. Steroids are an effective therapy for treating this condition.
| References | ▴Top |
- Pineda AA, Vamvakas EC, Gorden LD, Winters JL, Moore SB. Trends in the incidence of delayed hemolytic and delayed serologic transfusion reactions. Transfusion. 1999;39(10):1097-1103.
doi pubmed - Win N, New H, Lee E, de la Fuente J. Hyperhemolysis syndrome in sickle cell disease: case report (recurrent episode) and literature review. Transfusion. 2008;48(6):1231-1238.
doi pubmed - Morawakage LR, Perera BJ, Dias PD, Wijewardana SK. Hyperhemolysis in a patient with beta-thalassemia major. Asian J Transfus Sci. 2009;3(1):26-27.
doi pubmed - Darabi K, Dzik S. Hyperhemolysis syndrome in anemia of chronic disease. Transfusion. 2005;45(12):1930-1933.
doi pubmed - Treleaven JG, Win N. Hyperhaemolysis syndrome in a patient with myelofibrosis. Hematology. 2004;9(2):147-149.
doi pubmed - Petz LD, Calhoun L, Shulman IA, Johnson C, Herron RM. The sickle cell hemolytic transfusion reaction syndrome. Transfusion. 1997;37(4):382-392.
doi pubmed - Bachmeyer C, Maury J, Parrot A, Bachir D, Stankovic K, Girot R, Lionnet F. Rituximab as an effective treatment of hyperhemolysis syndrome in sickle cell anemia. Am J Hematol. 2010;85(1):91-92.
pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.