

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website http://www.thejh.org |
Review
Volume 5, Number 2, June 2016, pages 41-48
Potential Benefits of Metformin Use in Sickle Cell Anemia
Nejc Umeka, c, Chiedozie Kenneth Ugwokeb
aFaculty of Medicine, University of Ljubljana, Slovenia
bFaculty of Medical Sciences, University of Nigeria, Nigeria
cCorresponding Author: Nejc Umek, Faculty of Medicine, University of Ljubljana, Slovenia
Manuscript accepted for publication May 23, 2016
Short title: Metformin in Sickle Cell Anemia
doi: http://dx.doi.org/10.14740/jh275w
- Abstract
- Introduction
- Molecular Pharmacology of Metformin
- Effect of Metformin on Fetal Hemoglobin (HbF) Synthesis
- Effect of Metformin on Nitric Oxide (NO) Production
- Effect of Metformin on Inflammatory Phenotype
- Effect of Metformin on Ischemia/Reperfusion Injury
- Clinical Considerations for Metformin Use in SCA
- Conclusion
- References
| Abstract | ▴Top |
Sickle cell anemia (SCA) is one of the most important genetic disorders known to mankind. Even with the impressive advances in medical science, effective treatment and cure remain challenging. Currently available treatments including hydroxyurea, which is the only FDA approved drug for SCA, and hemopoietic stem cell transplantation all have significant limitations, so the search for new therapeutic options continues. Metformin, the traditional antidiabetic drug has recently gained novel attention from accumulating molecular evidence suggesting its pleiotropic effects and new potential applications. Our study of these new understandings of the pharmacodynamics and pleiotropic effects leads us to propose that the drug may be of potential therapeutic benefit in SCA. Our arguments are premised on a logical correlation of the interconnected pathophysiologic mechanisms in SCA with current information on the molecular pharmacodynamics of metformin. We reviewed existing evidence and deduced the diverse effects of metformin of relevant therapeutic significance in SCA, including enhancement of nitric oxide bioavailability, induction of fetal hemoglobin synthesis, attenuation of the inflammatory phenotype and beneficial effects in ischemia/reperfusion injury. Collectively, these considerations lead us to infer that there is reasonable evidence to support potential therapeutic adaptation of metformin in SCA.
Keywords: Metformin; Sickle cell anemia; Fetal hemoglobin; eNOS; Inflammation; Ischemia/reperfusion injury
| Introduction | ▴Top |
Sickle cell anemia (SCA) is one of the most prevalent genetic disorders in Sub-Saharan Africa, Middle East and India [1]. Approximately 5% of the world’s population carry a gene for hemoglobinopathies, mainly for sickle cell disease and thalassemias. Due to increased globalization and migration, there is an increasing gene flow from high gene frequency areas to Europe and USA. It is predicted that the global number of newborns with SCA will increase in the next 35 years from 305,800 in 2010 to 404,200 in 2050. Nigeria and Democratic Republic of Congo are and will probably remain the countries with highest burden of SCA [2]. Managing patients with SCA is a very high financial burden for their families and countries. In USA, the average lifetime cost of care is estimated to $460,151 per patient with SCA [3].
SCA is a qualitative genetic hemoglobinopathy caused by a single amino acid substitution in the beta globin chain of hemoglobin (Hb), which leads to expression of an abnormal protein called hemoglobin S (HbS). In its deoxygenated form, this abnormal HbS polymerizes and gives the erythrocytes their particular sickle shape. Such deformed cells are more fragile and prone to hemolysis, so the first clinical manifestation of the disease is usually severe hemolytic anemia [4]. Sickle blood cells are also less deformable and more adherent to the endothelium, which favors their entrapment in the microvasculature, resulting in local blood flow disturbances, vaso-occlusive crises and infarction of vital organs. Resolution of such vaso-occlusive episodes leads to oxidative stress and inflammation that contribute to vasculopathy [5]. Even with advanced medical care, including red blood cell transfusions and hydroxyurea, these vaso-occlusive events and vasculopathy usually result in central nervous system thrombosis, priapism, acute and chronic pain syndromes, retinopathy, pulmonary hypertension, heart failure, kidney failure, skin ulcerations and severe infections [6].
Since 1958, metformin has been one of the most commonly prescribed antihyperglycemic drugs for the treatment of type 2 diabetes mellitus [7]. Two decades ago, Verma et al [8] and Bhalla et al [9] noticed that metformin also has direct vascular effects in hypertensive rats. These effects were also supported by large-scale clinical trials that have demonstrated that metformin improves vascular function [10] and reduces mortality by actions that cannot be attributed entirely to its antihyperglycemic effects [11]. Since then, our understanding of molecular pharmacodynamics of metformin continues to expand with accumulating evidence, offering new interesting insights into potential clinical applications of the drug such as anti-cancer, anti-aging, anti-inflammatory agent, etc. [12]. Our study of the new understanding regarding the molecular pharmacodynamics of metformin has similarly led us to propose that the drug may be potentially adapted for therapeutic benefit in SCA. In the discussion that follows, we shall explore relevant aspects of the pathophysiology of SCA, and attempt to correlate how new insights in the molecular pharmacology of metformin may fit in for therapeutic exploitation.
| Molecular Pharmacology of Metformin | ▴Top |
Molecular mechanisms of metformin action are very complex and still partially unknown. Although the direct target of metformin remains unknown, its molecular pharmacodynamics is classically divided into AMP-activated protein kinase (AMPK) dependent and AMPK independent mechanisms [13]. AMPK is a heterotrimeric complex that is found in almost all eukaryotic cells where it acts as a sensor of cellular energy state [14]. It is mainly activated by AMP, which promotes the phosphorylation of AMPK by upstream kinases, like LKB1, and prevents its dephosphorylation by protein phosphatises [15]. In vitro models have shown that metformin inhibits mitochondrial complex I of respiratory chain, which lowers the production of ATP and thus increases AMP/ATP ratio [13]. At high concentrations, metformin inhibits AMP deaminase, which also increases AMP concentration [16]. Lately it was found that metformin also activates AMPK directly by promoting formation of the αβγ heterotrimeric complex [17]. When activated, AMPK inhibits anabolic pathways like triglyceride and protein synthesis and stimulates catabolic pathways like fatty acid oxidation and glycolysis [14]. Metformin also has many AMPK independent effects, the clinical importance of which is still not known [18], but the main one seems to be inhibition of mitochondrial glycerol-3-phosphate dehydrogenase which increases the cytosol NADH/NAD+ ratio and prevents utilization of glycerol and lactate [19] (Fig. 1).
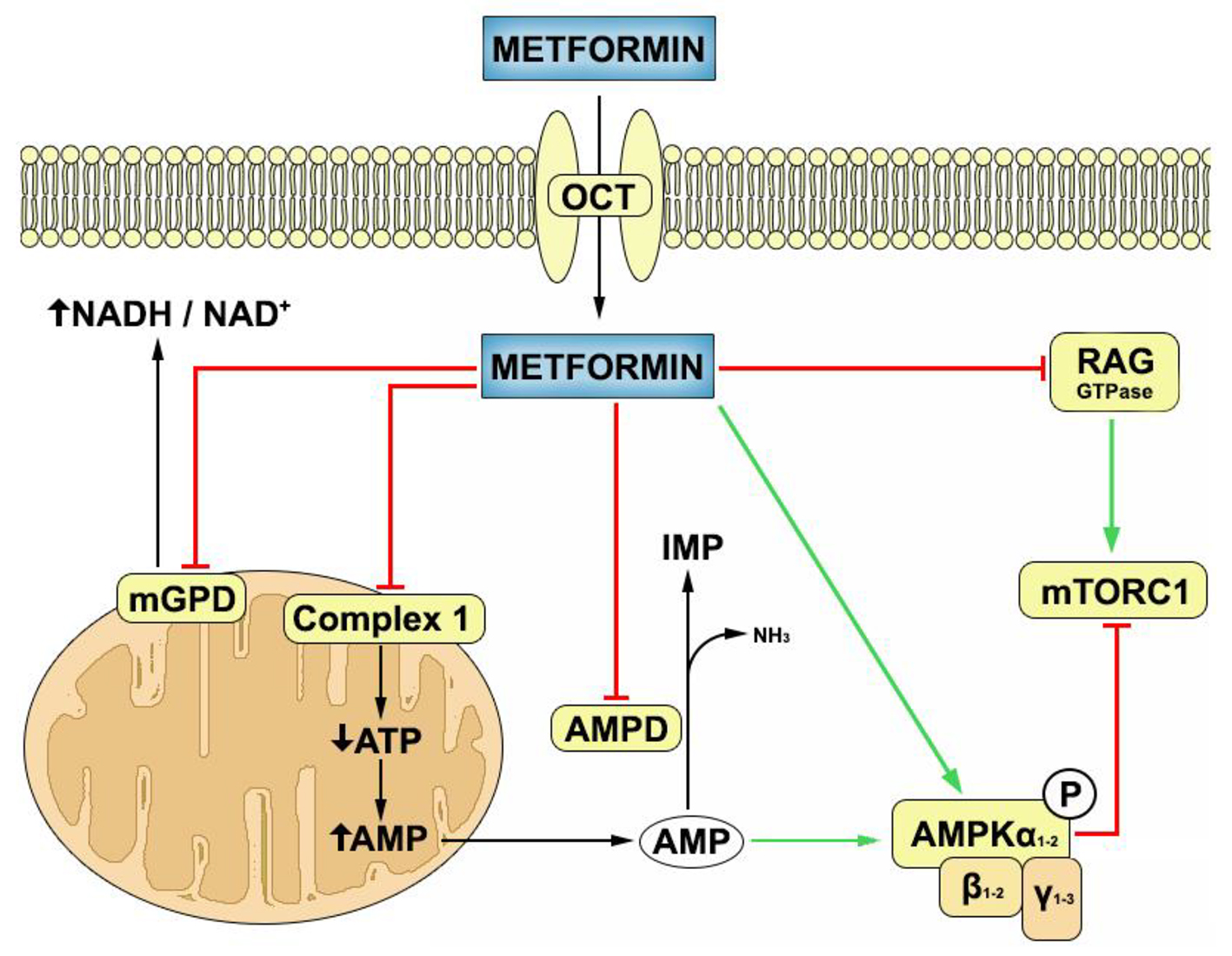 Click for large image | Figure 1. Mechanisms of metformin action inside the target cell. Metformin is transported into the cells through organic cation transporters (OCTs). Inside the cell, it inhibits complex 1 of mitochondrial respiratory chain, mitochondrial glycerol-3-phosphate dehydrogenase (mGPD) and AMP deaminase (AMPD), and stimulates the formation of AMPK heterotrimeric (αβγ) complex. Metformin can inhibit mammalian target of rapamycin complex 1 (mTORC1) through AMPK activation or through RAG GTPase inhibition. Green arrows denote stimulatory action while red arrows denote inhibitory action. |
At physiologic pH, metformin exists in cation form which makes passive diffusion through biologic membranes almost impossible [20]. It is transported in cells through organic cation transporters 1, 2 and 3 (OCT1, 2, 3) and multidrug and toxin extrusion 1 and 2-K (MATE1 and MATE2-K) which are extensively expressed in most human tissues and cell types [21].
| Effect of Metformin on Fetal Hemoglobin (HbF) Synthesis | ▴Top |
The induction of HbF synthesis is one of the primary targets of therapy in SCA [6]. It is known that the mammalian target of rapamycin (mTOR) signaling pathway is one of the key regulators of erythropoiesis [22, 23] and probably has a significant role in the pathobiology of SCA [24]. Inhibition of this pathway has been demonstrated to have a number of beneficial effects in SCA. As a case in point, rapamycin is a direct inhibitor of the mTOR pathway, and this inhibition has been shown to induce gamma-globin gene and increase HbF expression in erythroid precursor cells in vitro and in vivo [24, 25] and increase erythrocyte count and hemoglobin levels in beta-thalassemic mice models [22]. Rapamycin has also been demonstrated to ameliorate the pain phenotype in a humanized SCA mouse model, probably via a similar mTOR inhibition mechanism [24].
Metformin is known to inhibit mTOR complex 1 (mTORC1) signaling pathway via at least two different mechanisms. The first one is through activation of AMPK that phosphorylates tuberous sclerosis complex 2 (TSC2) and protein Raptor (regulatory associated protein of mTOR) which inhibit the mTORC1 [26], while the second mechanism is via upregulation of hexokinase II and inhibition of Rag GTPase [27, 28]. Furthermore, metformin as AMPK activator has been shown to induce and enhance the expression forkhead transcription factor 3 (FOXO3), a positive regulator of gamma-globin elaboration [29, 30], which is also involved in the erythroblast maturation process in cooperation with the mTOR pathway [22]. Since hemopoietic cells express organic cation transporter OCTN1 which is one of the transporters of metformin [31, 32], metformin may be of potential benefit in SCA possibly by an increase of HbF levels via mTOR inhibition and FOXO3 activation. Because metformin at therapeutic concentrations is a relatively weak mTOR inhibitor and FOXO3 activator in vivo, it is suggested that this potential of HbF elevation may be clinically significant only in the context of a synergistic effect with hydroxyurea or other HbF inducing agents in upregulation of gamma-globin synthesis [30].
| Effect of Metformin on Nitric Oxide (NO) Production | ▴Top |
NO is one of the most critical agents involved in the pathophysiology of sickle cell vasculopathy. In normal endothelial biology, NO plays key vaso-regulatory functions including acting as central mediator of endothelial-dependent vasodilatation, suppression of the procoagulant phenotype induction via inhibition of tissue factor expression, inhibition of platelet activation and downregulation of adhesion molecule elaboration, among others [33]. SCA is a chronic NO biodeficient state characterized by enhanced consumption and impaired generation of NO [34]. The mechanisms responsible for this NO biodeficiency profile are varied and include a hyper-hemolytic state which favors increased NO consumption by cell free hemoglobin, increased superoxide generation with enhanced NO scavenging and impaired endothelial NO synthase (eNOS) activity [33]. Although it is believed that in SCA eNOS enzyme levels may possibly be increased as an adaptive compensatory response [35], parallel evidence suggests that there is decreased bioavailability of NO from impairment of eNOS-NO system attributable to a number of mechanisms in the sickle context. These include increased levels of asymmetric dimethyl arginine (ADMA) [36], a potent inhibitor of eNOS; decreased ApoA-1 [33]; and uncoupling of eNOS due to arginine deficiency [37], relative insufficiency of tetrahydrobiopterin (THB4) [38] and eNOS oxidation and monomerization [39], with resultant generation of peroxynitrite rather than NO. The implications of this NO biodeficient profile in SCA pathophysiology thus include activation of a hypercoagulable state via inflammation-coagulation coupling, increased platelet activation, enhanced adhesion molecule expression, impaired endothelial-dependent vasodilatation and upregulation of proinflammatory pathways [33].
Several studies have demonstrated that metformin enhances the activity of the eNOS-NO system by increasing eNOS phosphorylation levels via AMPK [40-42]. Thus metformin by upregulating eNOS activity potentially inhibits the pathophysiologic consequences of NO biodeficiency in SCA. However, mindful that isolated upregulation of eNOS-NO system in the context of the uninhibited inflammatory milieu of SCA and relative THB4 and arginine deficiency [37, 38] may generate destructive superoxides rather than NO, metformin administration for therapeutic benefit should take into consideration concomitant supplementation of THB4 and arginine. Moreover, since hydroxyurea increases eNOS protein levels and potentiates NO generation [43], it may act synergistically with metformin in this regard. Furthermore, acute and chronic metformin treatments have been shown to foster improved revascularization following vaso-occlusive ischemia via AMPK-eNOS related mechanisms [40, 44]. In addition, metformin can significantly increase differentiation of endothelial progenitor cells (EPC) through AMPK-eNOS-NO and AMPK-mTOR-p70S6K pathways [45]. These cells originate from bone marrow [46] and have the capability of incorporating themselves into neovessels at the site of ischemia and secreting angiogenic growth factors [47] that can also contribute to revascularization. While a few agents that potentiate the eNOS-NO system have been explored for therapeutic exploitation, clinical adaptation of these agents has been unsuccessful on account of significant safety limitations. Inhaled NO is difficult to administer and needs to be intensively monitored [48], while sildenafil hitherto used to increase effects of endogenous NO has been discontinued due to serious side effects [49]. In this respect therefore, metformin may be more adapted for clinical application because of its relative proven safety profile [50].
| Effect of Metformin on Inflammatory Phenotype | ▴Top |
Inflammation biology is one of the central themes in the pathophysiology of the SCA, and much of the clinical events of the disease are consequent on the context of a chronic systemic inflammatory state. The inflammatory phenotype is mainly due to increased hemolysis and ischemia/reperfusion injury and is characterized by chronically elevated plasma levels of proinflammatory cytokines including tumor necrosis factor α (TNF-α), interleukin 6 (IL-6), interleukin 1-β (IL-1β), interleukin 17 (IL-17) and interleukin 8 (IL-8), and reduced level of the anti-inflammatory cytokine, interleukin 10 (IL-10) [51, 52]. There are also raised levels of other inflammatory markers like interferon γ (IF-γ), C-reactive protein (CRP), macrophage inflammatory protein 1α (MIP-1α) and monocyte chemotactic protein 1 (MCP-1) [53].
A growing body of evidence suggests that metformin may exert both direct and indirect anti-inflammatory effects beyond the established antihyperglycemic action [54]. A number of molecular mechanisms have been proposed for this anti-inflammatory effect including the inhibition of nuclear factor κB (NFκB) via AMPK dependent and independent pathways [55]; activation of AMPK-eNOS-NO pathway [54]; suppression of STAT3 signaling pathway and regulation of the balance between Th17/Treg [56]; inhibition of the stress responsive proteins SIRT 7 and SIRT 1 which may mediate inflammation [57]; and AMPK and poly(ADP ribose) polymerase 1 (PARP-1) dependent upregulation of B-cell lymphoma-6 protein (Bcl-6) which inhibits expression of proinflammatory mediators [58]. Furthermore, it is also suggested that metformin inhibits the formation of advanced glycation end products (AGEs) via direct reaction between metformin and dicarbonyl precursors of AGEs [59] and downregulates the expression of receptor for AGEs (RAGE) through AMPK activation [60], which reduces formation of ROS and inflammation [61].
Metformin has been reported to exert therapeutic anti-inflammatory effects in several inflammatory diseases including autoimmune arthritis [62], inflammatory bowel disease [56] and other autoimmune diseases [63] via decreased expression of inflammatory mediators like IL-1β, IL-6, IL-8, IL-2, TNF-α, MCP-1, CRP, IFN-γ, and upregulation of IL-10 [62, 64]. Therefore, metformin treatment may have therapeutic benefits in SCA via suppression of the inflammatory phenotype and consequent attenuation of the inflammation-dependent clinical course and manifestations of the disease such as chronic pain, lung injury and impaired cognitive function despite normal brain MRI [65, 66]. Furthermore, via suppression of endothelial activation and adhesion molecules expression, this potential amelioration of inflammation may also decrease the frequency and severity of acute vaso-occlusive events [33].
| Effect of Metformin on Ischemia/Reperfusion Injury | ▴Top |
Ischemia/reperfusion injury is a cardinal pathophysiologic mechanism of the inflammatory milieu and consequent vasculopathic complications of SCA in various organs (acute kidney injury, kidney failure, ischemic stroke, myocardial infarction, skin ulcerations, etc.). The cellular insults from ischemia/reperfusion injury are associated with oxidative stress and bioenergetic crisis [4]. A cellular response to ischemic stress is activation of AMPK, a key homeostatic energy sensor that preconditions favorable cellular bioenergetics and induces cellular survival mechanisms in stressful conditions [14]. This default cellular response is however insufficient to abort the cellular injury in ischemia/reperfusion. AMPK activators such as metformin therefore should potentially enhance cellular preconditioning for ischemic events and consequently enhance cellular tolerance and resilience to hypoxic stress. Indeed, accumulating evidence exists to support this logical inference. Several studies have reported that acute and chronic preconditioning with metformin ameliorates ischemia/reperfusion injury in various organs like the brain, kidney, heart, liver and skin. However, exact molecular mechanisms of these effects in various organs are yet to be clarified.
In the brain, it was demonstrated that both acute and chronic metformin preconditioning confers neurovascular protection against cerebral ischemia by pre-activation of brain AMPK and inhibition of NF-κB mediated inflammatory pathway [67, 68]. Metformin has also been noted to attenuate post-ischemic reactive hyperemia and neuronal excitation and promote angiogenesis and neurogenesis following cerebral ischemia [44, 69, 70]. Collectively, the evidence highlighted suggests a practical benefit of metformin in stroke prevention and amelioration of cerebral ischemic injury. This benefit is of potential significance in SCA where ischemic cerebrovascular accidents are a major cause of morbidity and mortality.
In the kidney, ischemia/reperfusion is a form of acute kidney injury and activation of AMPK has been shown to confer renal protection [71]. Metformin preconditioning has been shown to protect against renal tubular injury via attenuation of inflammation consequent on renal ischemia/reperfusion and inhibition of hypoxia inducible factor 1α (HIF-1α) accumulation in renal proximal tubular epithelial cells [72, 73]. Hence, metformin preconditioning may be potentially beneficial in preventing acute kidney injury due to ischemia/reperfusion and suppressing nephropathy progression in SCA patients.
In the heart, a number of studies have demonstrated that metformin treatment suppresses inflammatory response following cardiac ischemia via AMPK activation, and confers protective benefits in myocardial infarction [12, 74]. Also, metformin has been shown to protect against cardiac ischemia via Akt-PI3K dependent inhibition of mitochondrial permeability transition pore (mPTP) opening [75]. Therefore, metformin could also potentially ameliorate cardiac ischemic/reperfusion injury in SCA patients.
There are also few reports in literature of similar protective effects in other organs like the skin and liver. In the liver, metformin has been shown to prevent ischemic/reperfusion induced oxidative stress via increased antioxidant activity, and decreased ROS production and post-ischemic inflammation [76]. Foot ulcers are common complications of SCA vasculopathy often requiring plastic surgical intervention. Metformin pre-treatment has been shown to improve skin flap survival via NO-mediated mechanism [77].
Li et al demonstrated that ischemia/reperfusion inducible proteins (IRIP) negatively modulate the function of OCT1 and MATE1 in cells which may cause an alteration in metformin disposition in tissues after ischemia/reperfusion injury [78]. The clinical importance of such modulation is not known and needs to be characterized.
| Clinical Considerations for Metformin Use in SCA | ▴Top |
Given that metformin can act via at least four different major points in the interconnected pathophysiology of SCA, it is reasonable to suspect that overall clinical effect might be significant (Fig. 2). However, while there has been compelling evidence regarding its general safety profile in conventional clinical use [50], there is paucity of specific data on metformin safety in SCA. Indeed a number of adverse effects may be associated with metformin use, including reported risk of cognitive impairment, and a relatively low risk of lactic acidosis (4.3/100,000 patient-year) especially in the context of renal and hepatic failure [50, 54]. This may be important given that nephropathy and renal failure are common associated complications of SCA [6]. Hence, direct evidence is needed to clarify to what extent the peculiar renal and hepatic pathophysiology of SCA may limit metformin’s therapeutic adaptation. Moreover, metformin safety in pediatric subjects has not been overwhelmingly explored, and while there is not currently significant evidence questioning its general safety in children [79], stronger validation of its pediatric safety profile is needed before adaptation to clinical use in children who comprise the majority of SCA patients in third world countries [2]. Meanwhile, on a positive note metformin is generally well tolerated, does not cause hypoglycemia, and is a relatively affordable and available drug globally [7]. Overall therefore, further evidence is needed to clarify these concerns and objectively weigh the proposed benefits of metformin use in SCA against clinical considerations.
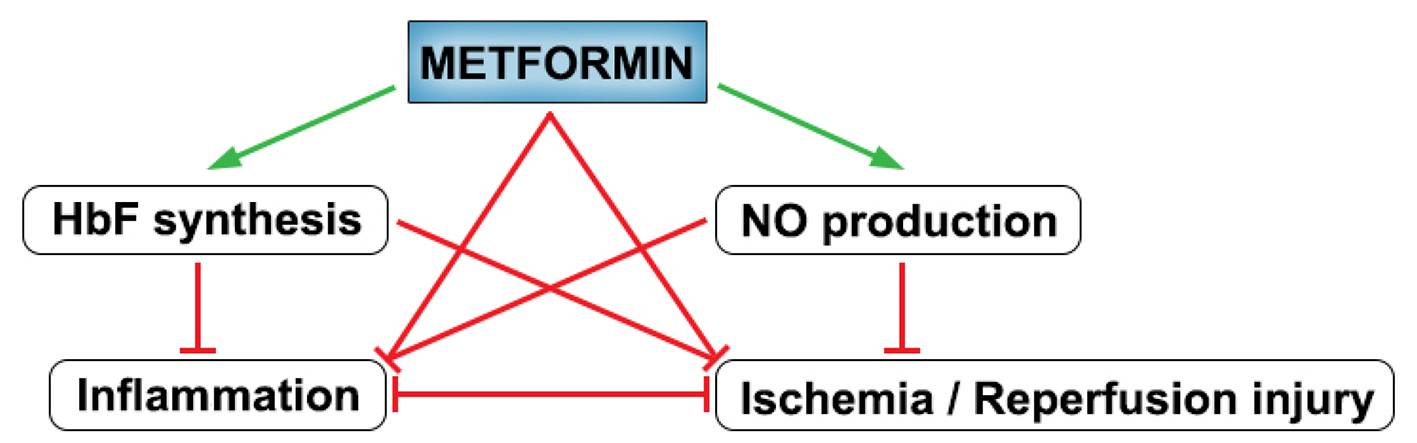 Click for large image | Figure 2. Summary of the effects of metformin on interconnected pathophysiologic processes in SCA. Green arrows denote stimulatory action while red arrows denote inhibitory action. |
| Conclusion | ▴Top |
In the present review, we have attempted to extend the spectrum of potential clinical utility of metformin by matching current molecular evidence with clinical relevance in SCA, and from the above considerations, we believe that there is significant deductive evidence to support potential clinical adaptation either as monotherapeutic agent or as an adjuvant to hydroxyurea. This potential is remarkable given the pleiotropic molecular pharmacodynamic effects of metformin on the various interrelated pathways and mechanisms in the pathophysiology of SCA. To the best of our knowledge, we are the first to comprehensively explore this pharmacodynamic/pathophysiologic correlation and advance a holistic molecular argument for the potential therapeutic exploitation of metformin in SCA. However, considering that the current evidence is largely derived from preclinical studies, we propose that more specific animal studies and subsequent human studies are needed to conclusively validate, modify or refute these arguments.
Acknowledgments
We are grateful to Dr. Gregor Vercek for editing the manuscript and to Urban Slokar for assistance with graphics.
Conflicts of Interest
The authors confirm that this article content has no conflicts of interest.
| References | ▴Top |
- Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86(6):480-487.
doi pubmed - Piel FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, Dewi M, Temperley WH, et al. Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. Lancet. 2013;381(9861):142-151.
doi - Kauf TL, Coates TD, Huazhi L, Mody-Patel N, Hartzema AG. The cost of health care for children and adults with sickle cell disease. Am J Hematol. 2009;84(6):323-327.
doi pubmed - Kaul DK, Fabry ME. In vivo studies of sickle red blood cells. Microcirculation. 2004;11(2):153-165.
pubmed - Chirico EN, Pialoux V. Role of oxidative stress in the pathogenesis of sickle cell disease. IUBMB Life. 2012;64(1):72-80.
doi pubmed - Frenette PS, Atweh GF. Sickle cell disease: old discoveries, new concepts, and future promise. J Clin Invest. 2007;117(4):850-858.
doi pubmed - Inzucchi SE, Bergenstal RM, Buse JB, Diamant M, Ferrannini E, Nauck M, Peters AL, et al. Management of hyperglycemia in type 2 diabetes, 2015: a patient-centered approach: update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2015;38(1):140-149.
doi pubmed - Verma S, Bhanot S, McNeill JH. Decreased vascular reactivity in metformin-treated fructose-hypertensive rats. Metabolism. 1996;45(9):1053-1055.
doi - Bhalla RC, Toth KF, Tan E, Bhatty RA, Mathias E, Sharma RV. Vascular effects of metformin. Possible mechanisms for its antihypertensive action in the spontaneously hypertensive rat. Am J Hypertens. 1996;9(6):570-576.
doi - Abbasi F, Chu JW, McLaughlin T, Lamendola C, Leary ET, Reaven GM. Effect of metformin treatment on multiple cardiovascular disease risk factors in patients with type 2 diabetes mellitus. Metabolism. 2004;53(2):159-164.
doi pubmed - Kirpichnikov D, McFarlane SI, Sowers JR. Metformin: an update. Ann Intern Med. 2002;137(1):25-33.
doi pubmed - Bromage DI, Yellon DM. The pleiotropic effects of metformin: time for prospective studies. Cardiovasc Diabetol. 2015;14:109.
doi pubmed - Gong L, Goswami S, Giacomini KM, Altman RB, Klein TE. Metformin pathways: pharmacokinetics and pharmacodynamics. Pharmacogenet Genomics. 2012;22(11):820-827.
doi pubmed - Hardie DG. AMPK - sensing energy while talking to other signaling pathways. Cell Metab. 2014;20(6):939-952.
doi pubmed - Gowans GJ, Hawley SA, Ross FA, Hardie DG. AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab. 2013;18(4):556-566.
doi pubmed - Ouyang J, Parakhia RA, Ochs RS. Metformin activates AMP kinase through inhibition of AMP deaminase. J Biol Chem. 2011;286(1):1-11.
doi pubmed - Meng S, Cao J, He Q, Xiong L, Chang E, Radovick S, Wondisford FE, et al. Metformin activates AMP-activated protein kinase by promoting formation of the alphabetagamma heterotrimeric complex. J Biol Chem. 2015;290(6):3793-3802.
doi pubmed - Turban S, Stretton C, Drouin O, Green CJ, Watson ML, Gray A, Ross F, et al. Defining the contribution of AMP-activated protein kinase (AMPK) and protein kinase C (PKC) in regulation of glucose uptake by metformin in skeletal muscle cells. J Biol Chem. 2012;287(24):20088-20099.
doi pubmed - Madiraju AK, Erion DM, Rahimi Y, Zhang XM, Braddock DT, Albright RA, Prigaro BJ, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014;510(7506):542-546.
doi pubmed - Graham GG, Punt J, Arora M, Day RO, Doogue MP, Duong JK, Furlong TJ, et al. Clinical pharmacokinetics of metformin. Clin Pharmacokinet. 2011;50(2):81-98.
doi pubmed - Koepsell H, Lips K, Volk C. Polyspecific organic cation transporters: structure, function, physiological roles, and biopharmaceutical implications. Pharm Res. 2007;24(7):1227-1251.
doi pubmed - Zhang X, Camprecios G, Rimmele P, Liang R, Yalcin S, Mungamuri SK, Barminko J, et al. FOXO3-mTOR metabolic cooperation in the regulation of erythroid cell maturation and homeostasis. Am J Hematol. 2014;89(10):954-963.
doi pubmed - Knight ZA, Schmidt SF, Birsoy K, Tan K, Friedman JM. A critical role for mTORC1 in erythropoiesis and anemia. Elife. 2014;3:e01913.
doi pubmed - Khaibullina A, Almeida LE, Wang L, Kamimura S, Wong EC, Nouraie M, Maric I, et al. Rapamycin increases fetal hemoglobin and ameliorates the nociception phenotype in sickle cell mice. Blood Cells Mol Dis. 2015;55(4):363-372.
doi pubmed - Mischiati C, Sereni A, Lampronti I, Bianchi N, Borgatti M, Prus E, Fibach E, et al. Rapamycin-mediated induction of gamma-globin mRNA accumulation in human erythroid cells. Br J Haematol. 2004;126(4):612-621.
doi pubmed - Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13(9):1016-1023.
doi pubmed - Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007;67(22):10804-10812.
doi pubmed - Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320(5882):1496-1501.
doi pubmed - Sato A, Sunayama J, Okada M, Watanabe E, Seino S, Shibuya K, Suzuki K, et al. Glioma-initiating cell elimination by metformin activation of FOXO3 via AMPK. Stem Cells Transl Med. 2012;1(11):811-824.
doi pubmed - Zhang Y, Crosby JR, Boerwinkle E, Pace B, Sheehan VA. Pharmacological Induction of FOXO3 Is a Potential Treatment for Sickle Cell Disease. Blood. 2015;126(23):282.
- Kobayashi D, Aizawa S, Maeda T, Tsuboi I, Yabuuchi H, Nezu J, Tsuji A, et al. Expression of organic cation transporter OCTN1 in hematopoietic cells during erythroid differentiation. Exp Hematol. 2004;32(12):1156-1162.
doi pubmed - Nakamichi N, Shima H, Asano S, Ishimoto T, Sugiura T, Matsubara K, Kusuhara H, et al. Involvement of carnitine/organic cation transporter OCTN1/SLC22A4 in gastrointestinal absorption of metformin. J Pharm Sci. 2013;102(9):3407-3417.
doi pubmed - Hebbel RP, Vercellotti G, Nath KA. A systems biology consideration of the vasculopathy of sickle cell anemia: the need for multi-modality chemo-prophylaxsis. Cardiovasc Hematol Disord Drug Targets. 2009;9(4):271-292.
doi pubmed - Reiter CD, Wang X, Tanus-Santos JE, Hogg N, Cannon RO, 3rd, Schechter AN, Gladwin MT. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med. 2002;8(12):1383-1389.
doi pubmed - Kaul DK, Liu XD, Fabry ME, Nagel RL. Impaired nitric oxide-mediated vasodilation in transgenic sickle mouse. Am J Physiol Heart Circ Physiol. 2000;278(6):H1799-1806.
pubmed - Schnog JB, Teerlink T, van der Dijs FP, Duits AJ, Muskiet FA. Plasma levels of asymmetric dimethylarginine (ADMA), an endogenous nitric oxide synthase inhibitor, are elevated in sickle cell disease. Ann Hematol. 2005;84(5):282-286.
doi pubmed - Morris CR, Kuypers FA, Larkin S, Vichinsky EP, Styles LA. Patterns of arginine and nitric oxide in patients with sickle cell disease with vaso-occlusive crisis and acute chest syndrome. J Pediatr Hematol Oncol. 2000;22(6):515-520.
doi pubmed - Katusic ZS, d'Uscio LV, Nath KA. Vascular protection by tetrahydrobiopterin: progress and therapeutic prospects. Trends Pharmacol Sci. 2009;30(1):48-54.
doi pubmed - Hsu LL, Champion HC, Campbell-Lee SA, Bivalacqua TJ, Manci EA, Diwan BA, Schimel DM, et al. Hemolysis in sickle cell mice causes pulmonary hypertension due to global impairment in nitric oxide bioavailability. Blood. 2007;109(7):3088-3098.
pubmed - Takahashi N, Shibata R, Ouchi N, Sugimoto M, Murohara T, Komori K. Metformin stimulates ischemia-induced revascularization through an eNOS dependent pathway in the ischemic hindlimb mice model. J Vasc Surg. 2015;61(2):489-496.
doi pubmed - Chen H, Li J, Yang O, Kong J, Lin G. Effect of metformin on insulin-resistant endothelial cell function. Oncol Lett. 2015;9(3):1149-1153.
doi - Ghosh S, Lakshmanan AP, Hwang MJ, Kubba H, Mushannen A, Triggle CR, Ding H. Metformin improves endothelial function in aortic tissue and microvascular endothelial cells subjected to diabetic hyperglycaemic conditions. Biochem Pharmacol. 2015;98(3):412-421.
doi pubmed - Cokic VP, Beleslin-Cokic BB, Noguchi CT, Schechter AN. Hydroxyurea increases eNOS protein levels through inhibition of proteasome activity. Nitric Oxide. 2007;16(3):371-378.
doi pubmed - Venna VR, Li J, Hammond MD, Mancini NS, McCullough LD. Chronic metformin treatment improves post-stroke angiogenesis and recovery after experimental stroke. Eur J Neurosci. 2014;39(12):2129-2138.
doi pubmed - Li WD, Du XL, Qian AM, Hu N, Kong LS, Wei S, Li CL, et al. Metformin regulates differentiation of bone marrow-derived endothelial progenitor cells via multiple mechanisms. Biochem Biophys Res Commun. 2015;465(4):803-809.
doi pubmed - Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, Witzenbichler B, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275(5302):964-967.
doi pubmed - Urbich C, Aicher A, Heeschen C, Dernbach E, Hofmann WK, Zeiher AM, Dimmeler S. Soluble factors released by endothelial progenitor cells promote migration of endothelial cells and cardiac resident progenitor cells. J Mol Cell Cardiol. 2005;39(5):733-742.
doi pubmed - Morris CR, Kuypers FA, Larkin S, Sweeters N, Simon J, Vichinsky EP, Styles LA. Arginine therapy: a novel strategy to induce nitric oxide production in sickle cell disease. Br J Haematol. 2000;111(2):498-500.
doi pubmed - Machado RF, Barst RJ, Yovetich NA, Hassell KL, Kato GJ, Gordeuk VR, Gibbs JS, et al. Hospitalization for pain in patients with sickle cell disease treated with sildenafil for elevated TRV and low exercise capacity. Blood. 2011;118(4):855-864.
doi pubmed - Jang K, Chung H, Yoon JS, Moon SJ, Yoon SH, Yu KS, Kim K, et al. Pharmacokinetics, Safety, and Tolerability of Metformin in Healthy Elderly Subjects. J Clin Pharmacol. 2015.
doi pubmed - Bowers AS, Reid HL, Greenidge A, Landis C, Reid M. Blood viscosity and the expression of inflammatory and adhesion markers in homozygous sickle cell disease subjects with chronic leg ulcers. PLoS One. 2013;8(7):e68929.
doi pubmed - Ferreira SB, de Brito LC, Oliveira MP, Maciel KF, Martelli Junior H, Vieira LQ, Sobrinho AP. Periapical cytokine expression in sickle cell disease. J Endod. 2015;41(3):358-362.
doi pubmed - Qari MH, Dier U, Mousa SA. Biomarkers of inflammation, growth factor, and coagulation activation in patients with sickle cell disease. Clin Appl Thromb Hemost. 2012;18(2):195-200.
doi pubmed - Saisho Y. Metformin and Inflammation: Its Potential Beyond Glucose-lowering Effect. Endocr Metab Immune Disord Drug Targets. 2015;15(3):196-205.
doi pubmed - Lu J, Ji J, Meng H, Wang D, Jiang B, Liu L, Randell E, et al. The protective effect and underlying mechanism of metformin on neointima formation in fructose-induced insulin resistant rats. Cardiovasc Diabetol. 2013;12:58.
doi pubmed - Lee SY, Lee SH, Yang EJ, Kim EK, Kim JK, Shin DY, Cho ML. Metformin Ameliorates Inflammatory Bowel Disease by Suppression of the STAT3 Signaling Pathway and Regulation of the between Th17/Treg Balance. PLoS One. 2015;10(9):e0135858.
doi pubmed - Aljada A. Lipopolysaccharides-Induced Inflammatory Response in White Blood Cells Is Associated with Alterations in Senescence Mediators: Modulation by Metformin. Metab Syndr Relat Disord. 2015;13(6):278-285.
doi pubmed - Gongol B, Marin T, Peng IC, Woo B, Martin M, King S, Sun W, et al. AMPKalpha2 exerts its anti-inflammatory effects through PARP-1 and Bcl-6. Proc Natl Acad Sci U S A. 2013;110(8):3161-3166.
doi pubmed - Beisswenger P, Ruggiero-Lopez D. Metformin inhibition of glycation processes. Diabetes Metab. 2003;29(4 Pt 2):6S95-103.
- Ishibashi Y, Matsui T, Takeuchi M, Yamagishi S. Metformin inhibits advanced glycation end products (AGEs)-induced renal tubular cell injury by suppressing reactive oxygen species generation via reducing receptor for AGEs (RAGE) expression. Horm Metab Res. 2012;44(12):891-895.
doi pubmed - Ramasamy R, Yan SF, Schmidt AM. The diverse ligand repertoire of the receptor for advanced glycation endproducts and pathways to the complications of diabetes. Vascul Pharmacol. 2012;57(5-6):160-167.
doi pubmed - Yan H, Zhou HF, Hu Y, Pham CT. Suppression of experimental arthritis through AMP-activated protein kinase activation and autophagy modulation. J Rheum Dis Treat. 2015;1(1):5.
pubmed - Tomczynska M, Bijak M, Saluk J. Metformin - the drug for the treatment of autoimmune diseases; a new use of a known anti-diabetic drug. Curr Top Med Chem. 2016.
doi pubmed - Hyun B, Shin S, Lee A, Lee S, Song Y, Ha NJ, Cho KH, et al. Metformin Down-regulates TNF-alpha Secretion via Suppression of Scavenger Receptors in Macrophages. Immune Netw. 2013;13(4):123-132.
doi pubmed - Maitre B, Habibi A, Roudot-Thoraval F, Bachir D, Belghiti DD, Galacteros F, Godeau B. Acute chest syndrome in adults with sickle cell disease. Chest. 2000;117(5):1386-1392.
doi pubmed - Andreotti C, King AA, Macy E, Compas BE, DeBaun MR. The Association of Cytokine Levels With Cognitive Function in Children With Sickle Cell Disease and Normal MRI Studies of the Brain. J Child Neurol. 2015;30(10):1349-1353.
doi pubmed - Jiang T, Yu JT, Zhu XC, Wang HF, Tan MS, Cao L, Zhang QQ, et al. Acute metformin preconditioning confers neuroprotection against focal cerebral ischaemia by pre-activation of AMPK-dependent autophagy. Br J Pharmacol. 2014;171(13):3146-3157.
doi pubmed - Zhu XC, Jiang T, Zhang QQ, Cao L, Tan MS, Wang HF, Ding ZZ, et al. Chronic Metformin Preconditioning Provides Neuroprotection via Suppression of NF-kappaB-Mediated Inflammatory Pathway in Rats with Permanent Cerebral Ischemia. Mol Neurobiol. 2015;52(1):375-385.
doi pubmed - Farbood Y, Sarkaki A, Khalaj L, Khodagholi F, Badavi M, Ashabi G. Targeting Adenosine Monophosphate-Activated Protein Kinase by Metformin Adjusts Post-Ischemic Hyperemia and Extracellular Neuronal Discharge in Transient Global Cerebral Ischemia. Microcirculation. 2015;22(7):534-541.
doi pubmed - Liu Y, Tang G, Zhang Z, Wang Y, Yang GY. Metformin promotes focal angiogenesis and neurogenesis in mice following middle cerebral artery occlusion. Neurosci Lett. 2014;579:46-51.
doi pubmed - Seo-Mayer PW, Thulin G, Zhang L, Alves DS, Ardito T, Kashgarian M, Caplan MJ. Preactivation of AMPK by metformin may ameliorate the epithelial cell damage caused by renal ischemia. Am J Physiol Renal Physiol. 2011;301(6):F1346-1357.
doi pubmed - Wang ZS, Liu XH, Wang M, Jiang GJ, Qiu T, Chen ZY, Wang L. Metformin attenuated the inflammation after renal ischemia/reperfusion and suppressed apoptosis of renal tubular epithelial cell in rats. Acta Cir Bras. 2015;30(9):617-623.
doi pubmed - Takiyama Y, Harumi T, Watanabe J, Fujita Y, Honjo J, Shimizu N, Makino Y, et al. Tubular injury in a rat model of type 2 diabetes is prevented by metformin: a possible role of HIF-1alpha expression and oxygen metabolism. Diabetes. 2011;60(3):981-992.
doi pubmed - El Messaoudi S, Rongen GA, de Boer RA, Riksen NP. The cardioprotective effects of metformin. Curr Opin Lipidol. 2011;22(6):445-453.
doi pubmed - Bhamra GS, Hausenloy DJ, Davidson SM, Carr RD, Paiva M, Wynne AM, Mocanu MM, et al. Metformin protects the ischemic heart by the Akt-mediated inhibition of mitochondrial permeability transition pore opening. Basic Res Cardiol. 2008;103(3):274-284.
doi pubmed - Cahova M, Palenickova E, Dankova H, Sticova E, Burian M, Drahota Z, Cervinkova Z, et al. Metformin prevents ischemia reperfusion-induced oxidative stress in the fatty liver by attenuation of reactive oxygen species formation. Am J Physiol Gastrointest Liver Physiol. 2015;309(2):G100-111.
doi pubmed - Taleb S, Moghaddas P, Rahimi Balaei M, Rahimpour S, Abbasi A, Ejtemaei-Mehr S, Dehpour AR. Metformin improves skin flap survival through nitric oxide system. J Surg Res. 2014;192(2):686-691.
doi pubmed - Li Q, Yang H, Peng X, Guo D, Dong Z, Polli JE, Shu Y. Ischemia/Reperfusion-inducible protein modulates the function of organic cation transporter 1 and multidrug and toxin extrusion 1. Mol Pharm. 2013;10(7):2578-2587.
doi pubmed - Marques P, Limbert C, Oliveira L, Santos MI, Lopes L. Metformin effectiveness and safety in the management of overweight/obese nondiabetic children and adolescents: metabolic benefits of the continuous exposure to metformin at 12 and 24 months. Int J Adolesc Med Health. 2016.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.