

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website http://www.thejh.org |
Original Article
Volume 6, Number 1, March 2017, pages 12-20
Warm Autoimmune Hemolytic Anemia: Clinical Profile and Management
Sreenivasa Rao Sudulaguntaa, f, Monica Kumbhatb, Mahesh Babu Sodalaguntac, Aravinda Settikere Natarajud, Shiva Kumar Bangalore Rajae, Keshava Chandra Thejaswie, Raj Deepake, Asif Hussain Mohammede, Sony P. Sunnye, Amulya Viswesware, Mikita Suvarnae, Rashmi Nanjappae
aDepartment of Internal Medicine, Dr.B.R.Ambedkar Medical College, Bangalore 560032, India
bDepartment of Pathology, Sri Ramachandra Medical College, Chennai, India
cDepartment of General Medicine, KS Hegde Medical College, Karnataka, India
dDepartment of Internal Medicine, Medical Services, Columbia Asia Hospital, Hebbal, Bangalore, India
eDepartment of General Medicine, Dr.B.R.Ambedkar Medical College, Bangalore, India
fCorresponding Author: Sreenivasa Rao Sudulagunta, Department of Internal Medicine, Dr.B.R.Ambedkar Medical College, K.G.Halli, Bangalore 560032, India
Manuscript accepted for publication November 04, 2017
Short title: Warm Autoimmune Hemolytic Anemia
doi: https://doi.org/10.14740/jh303w
| Abstract | ▴Top |
Background: Autoimmune hemolytic anemia (AIHA) is a rare autoimmune disease in which autoantibodies target red blood cells leading to marked decrease in their lifespan. The classification of AIHA is based on the immunochemical properties of the RBC autoantibody. Warm antibody AIHA (wAIHA) accounts for 75-80% of all adult AIHA cases. The treatment of wAIHA is mainly corticosteroids. Our retrospective study aimed to study the clinical profile and management of wAIHA.
Methods: Data of 75 patients admitted with wAIHA or presented to outpatient department (previous medical records) with wAIHA between January 2003 and January 2016 were analyzed.
Results: In our study, females constituted 12 and 26 patients of primary and secondary wAIHA, while males constituted 17 and 20 patients of primary and secondary wAIHA, respectively. Mean hemoglobin level at AIHA onset was found to be 7.1 ± 1.7 g/dL in primary wAIHA group and 6.3 ± 1.2 g/dL in secondary wAIHA group, which is statistically significant. Splenectomy was used as mode of treatment in one (3.4%) patient of primary wAIHA group and 15 (32.60%) patients of secondary wAIHA group, which is statistically significant. Mean age of wAIHA onset was 69.7 ± 21.5 years in wAIHA group secondary to lymphoma and 54.3 ± 25.7 years in other wAIHA group, which is statistically significant.
Conclusion: The most common causes of secondary wAIHA are B-cell lymphoma, systemic lupus erythematosus, rheumatoid arthritis, chronic lymphocytic leukemia (CLL), common variable immune deficiency, renal cell carcinoma and secondary to drug usage (alpha methyldopa and carbamazepine), respectively. Reducing the cumulative dose of corticosteroids with second line treatment whenever possible and therefore reducing the risk of sepsis, specifically in older patients with comorbidities will reduce morbidity and mortality.
Keywords: Warm antibody autoimmune hemolytic anemia; Rituximab; Corticosteroids; Complete response; Partial response; Direct antiglobulin test; Prednisone
| Introduction | ▴Top |
Hemolysis means the destruction of erythrocytes prematurely. A hemolytic anemia will develop if erythrocyte loss cannot be compensated by bone marrow. The severity depends on the onset of hemolysis (gradual or abrupt) and the extent of erythrocyte destruction. Mild hemolysis usually is asymptomatic while severe hemolysis can be life threatening and can cause cardiopulmonary decompensation. Autoimmune hemolytic anemia (AIHA) and hereditary spherocytosis are due to extravascular hemolysis because the RBCs are destroyed in the spleen and other reticuloendothelial tissues [1].
Hemolytic anemia occurs due to many causes [2-12]. AIHA occurs due to warm or cold autoantibody types and, rarely, mixed type [13, 14]. In a recent population-based study [15], the incidence was found to be 0.8/100,000/year, but the prevalence is found to be 17/100,000 [16]. Warm antibody autoimmune hemolytic anemia (wAIHA) is the most common of the autoimmune hemolytic diseases [17].
AIHA can affect children (usually below age of 5) and adults, with an annual incidence of 1 - 3 per 100,000 individuals [18, 19]. The classification of AIHA is based on the immunochemical properties of the RBC autoantibody [20, 21]. WAIHA accounts for about 75-80% of all adult AIHA cases [18, 20]. AIHA is subdivided into primary (or idiopathic) and secondary depending on the presence of an associated and potentially causative disorder. In previous studies, many diseases and conditions were found to be associated with wAIHA, and secondary forms of wAIHA are considered to represent about 50% of all wAIHA cases [22].
The diagnosis of AIHA is based on the following laboratory findings: normocytic or macrocytic anemia, reticulocytosis, low serum haptoglobin levels, elevated lactate dehydrogenase (LDH) level, increased indirect bilirubin level, and a positive direct antiglobulin test (DAT) with a broad-spectrum antibody against immunoglobulin and complement (Fig. 1). Secondary cases may not have all of the typical laboratory findings of AIHA [23]. Two questions required for are treatment decision: 1) The type of the antibody involved? 2) Is it primary or secondary AIHA?
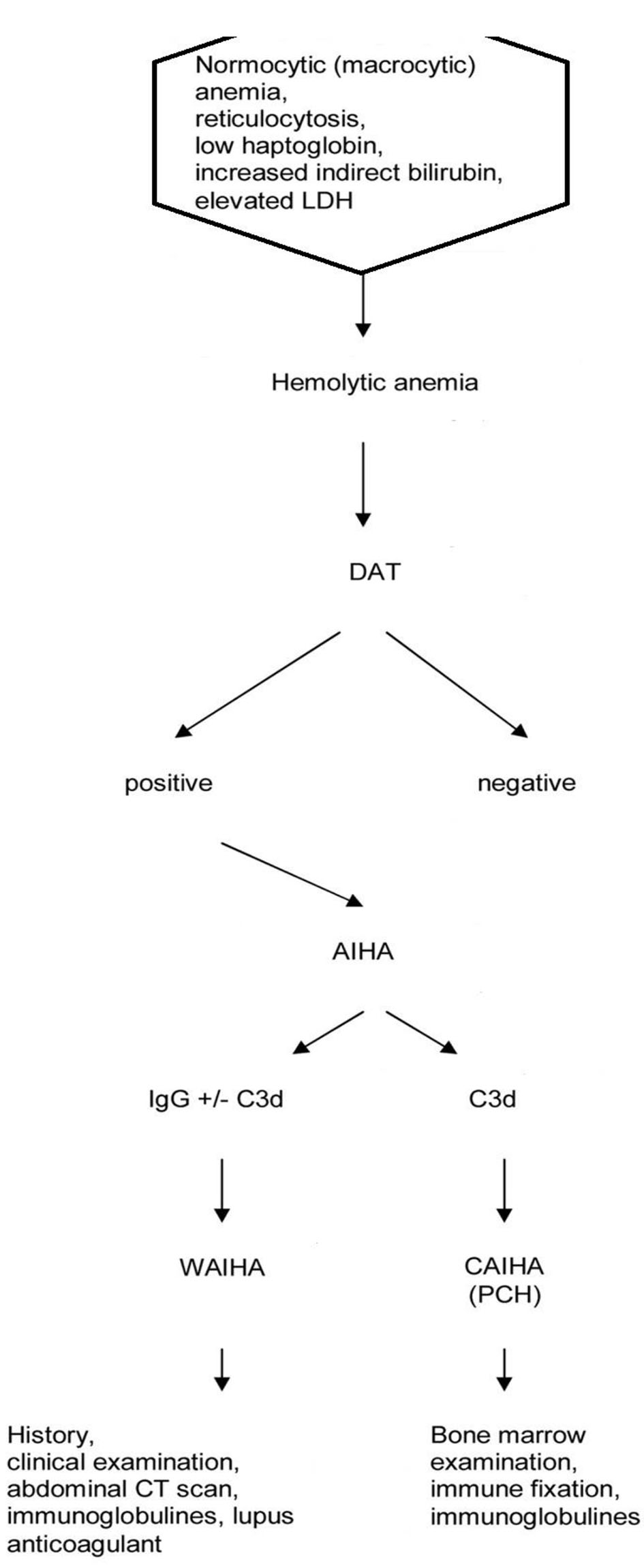 Click for large image | Figure 1. Flow chart of diagnosis of autoimmune hemolytic anemia. |
The type of antibody is identified with the use of monospecific antibodies to immunoglobulin G (IgG) and C3d. When the RBCs are coated with IgG or IgG plus C3d, the antibody is mostly a warm antibody (wAIHA). When the RBCs are coated with C3d only, the antibody is often, but not always a cold antibody. For definite diagnosis of a cold antibody AIHA (cAIHA), the cold agglutinin titer should be elevated (> 1:512). Diagnosis may be difficult in some patients with IgM warm antibodies, cold antibodies with low titers, DAT negativity, or Donath-Landsteiner antibodies. The type of antibodies and diseases underlying are represented in Table 1 [24-38].
 Click to view | Table 1. Prevalence and Type of Antibodies in Secondary AIHA in Adults |
The first line of treatment of newly diagnosed primary wAIHA is glucocorticoids. An initial dose of 1 mg/kg/day prednisone (PDN) is given orally or methylprednisolone intravenously. This initial dose is administered until the hematocrit reaches greater than 30% or a hemoglobin (Hb) level above 10 g/dL is reached. Normalization of Hb is not required. If the goal described above is not attained in 3 weeks, second-line treatment is started as further response with glucocorticoid treatment is not likely [39]. Once the treatment goal is attained, the dose of PDN is tapered to 20 - 30 mg/day within a few weeks.
Further dose of PDN is tapered slowly (by 2.5 - 5 mg/day per month) with monitoring of Hb and reticulocyte counts. An alternate-day regimen may further reduce the adverse effects of steroids. If the patient is in remission even after 3 - 4 months at a dose of 5 mg of PDN per day, an attempt to withdraw steroids is done. All patients on steroid therapy should receive vitamin D, bisphosphonates, and calcium from the beginning according to the recommendation of the American College of Rheumatology (ACR). Supplementation of folic acid is also recommended.
This retrospective study which evaluated 75 patients was conducted to study clinical profile, epidemiological and laboratory features of patients with wAIHA and mode of management, complications and prognostic factors.
| Methods | ▴Top |
The retrospective study analyzed data of patients admitted with wAIHA or presented to outpatient department (previous medical records) with wAIHA between January 2002 and January 2016. Data were pooled from seven hospitals. The medical records were analyzed for the demographic data (age and sex), clinical features, co-morbid conditions, investigations, mode and results of the treatment and complications of the procedures. A total of 81 patients fulfilled the clinical and diagnostic criteria of wAIHA of which 75 patients met the diagnostic certainty for wAIHA.
The eligibility criteria include the following: 1) age ≥ 16 years at the time of diagnosis; 2) diagnosis of AIHA defined by an Hb level ≤ 11 g/dL with laboratory features suggestive of hemolysis (low haptoglobin level and/or elevated LDH level and/or elevated bilirubin level) and a positive DAT result with an IgG or IgG1 C3d pattern; 3) absence of any other cause of hereditary or acquired hemolytic anemia. Patients with a history of Evans’ syndrome defined by sequential or simultaneous immune thrombocytopenia (ITP) [40] were included if the above criteria for wAIHA were fulfilled. Patients with C3d type only positive DAT result due to the presence of cold agglutinins were excluded. The diagnosis of other associated autoimmune disease was based on the ACR revised classification criteria for systemic lupus erythematosus (SLE), updated international criteria for definite primary antiphospholipid syndrome [41], and the European criteria for primary Sjogren syndrome [42].
Treatment efficacy was assessed based on the following criteria. A complete response (CR) was defined as Hb level ≥ 12 g/dL without blood transfusion recently and without hemolysis features (normal levels of bilirubin, LDH, and haptoglobin). A partial response (PR) was defined as Hb level ≥ 10 g/dL with an increase of minimum of 2 g from baseline and persistent hemolysis. At the end of the follow-up, complete remission was defined as a lasting CR without any treatment and partial remission as the need for a daily dose of PDN less than 10 mg to maintain at least a long-term remission. Corticosteroid dependency was defined as the need for long-term PDN to maintain at least a response or as early relapse (within 3 months) after withdrawal of PDN. In every other situation, patients were considered non-responders and/or as having active disease.
Statistical analyses were performed using SPSS 16 program. Data analyses were performed using Shapiro-Wilk tests, Chi-square tests, Fischer’s exact tests, and Wilcoxon two-sample tests on SAS and Graph Pad. P-value was considered significant if < 0.05. The study was performed in accordance with the ethical guidelines of the Helsinki Declaration and was approved by our ethics committee.
| Results | ▴Top |
In our study, females constituted 12 and 26 patients of primary and secondary wAIHA, while males constituted 17 and 20 patients of primary and secondary wAIHA, respectively (P > 0.05). The common clinical characteristics of primary and secondary wAIHA patients are represented in Table 2. Mean age of wAIHA onset was 51.7 ± 20.5 years in primary wAIHA group and 54.3 ± 25.7 years in secondary wAIHA, which is statistically not significant. Regarding clinical symptoms at the time of onset, 93.10% (27) in primary wAIHA group and 67.39% (31) in secondary wAIHA group reported it.
Click to view | Table 2. Comparison of Characteristics of Patients With Primary and Secondary wAIHA |
Anemia was diagnosed in 72.41% (21) of primary wAIHA group and 63.04% (29) of secondary wAIHA group. Jaundice or discolored urine was found in 37.93% (11) of primary wAIHA group and 32.60% (15) of secondary wAIHA group. Regarding chest pain or acute coronary syndrome at the time of onset, 10.34% (three) in primary wAIHA group and 10.86% (five) in secondary wAIHA group reported it. Mean Hb level at AIHA onset was found to be 7.1 ± 1.7 g/dL in primary wAIHA group and 6.3 ± 1.2 g/dL in secondary wAIHA group, which is statistically significant.
Mean reticulocyte level at AIHA onset was found to be 323 ± 179 × 109/L in primary wAIHA group and 262 ± 156 × 109/L in secondary wAIHA group, which is statistically insignificant. Mean corpuscular volume (MCV) level at AIHA onset was found to be 109 ± 16 fL in primary wAIHA group and 104 ± 18 fL in secondary wAIHA group, which is statistically insignificant. Decreased level of haptoglobin was found in 27 (93.10%) patients of primary wAIHA group and 43 (93.47%) patients of secondary wAIHA group, which is statistically insignificant.
Increased LDH level was found in 29 (100%) patients of primary wAIHA group and 42 (91.30%) patients of secondary wAIHA group, which is statistically insignificant. Increased bilirubin level was found in 25 (86.20%) patients of primary wAIHA group and 39 (84.78%) patients of secondary wAIHA group, which is statistically insignificant. Regarding DAT pattern, IgG antibody was found in 15 (51.72%) patients of primary wAIHA group and 15 (32.60%) patients of secondary wAIHA group, which is statistically insignificant.
IgG antibodies and complement (C3d) were found in 14 (48.27%) patients of primary wAIHA group and 30 (65.21%) patients of secondary wAIHA group, which is statistically insignificant. Only C3d was found in 0% patients of primary wAIHA group and one (2.17%) patient of secondary wAIHA group, which is statistically insignificant. IgA antibody was found in 0% patients of primary wAIHA group and two (4.34%) patients of secondary wAIHA group, which is statistically insignificant. Regarding the treatment given to wAIHA patients, blood transfusion was administered in 20 (68.96%) patients of primary wAIHA group and 34 (73.91%) patients of secondary wAIHA group, which is statistically insignificant.
Response to corticosteroid was observed in 28 (96.55%) patients of primary wAIHA group and 40 (86.95%) patients of secondary wAIHA group, which is statistically insignificant. Dependence on corticosteroid was observed in 17 (58.62%) patients of primary wAIHA group and 32 (69.56%) patients of secondary wAIHA group, which is statistically insignificant. CR to corticosteroid was observed in 20 (68.96%) patients of primary wAIHA group and 30 (65.21%) patients of secondary wAIHA group, which is statistically insignificant. Second-line treatment was administered in 19 (65.51%) patients of primary wAIHA group and 32 (69.56%) patients of secondary wAIHA group, which is statistically insignificant.
Rituximab was administered in 14 (48.27%) patients of primary wAIHA group and 23 (50%) patients of secondary wAIHA group, which is statistically insignificant. Splenectomy was used as mode of treatment in one (3.4%) patient of primary wAIHA group and 15 (32.60%) patients of secondary wAIHA group, which is statistically significant. Disease remission at last consultation was observed in 22 (75.86%) patients of primary wAIHA group and 33 (71.76%) patients of secondary wAIHA group, which is statistically insignificant. Complete remission of AIHA was observed in 14 (48.27%) patients of primary wAIHA group and 23 (50%) patients of secondary wAIHA group, which is statistically insignificant.
Partial remission of AIHA was observed in eight (27.58%) patients of primary wAIHA group and 12 (26.08%) patients of secondary wAIHA group, which is statistically insignificant. Active disease was observed in eight (27.58%) patients of primary wAIHA group and 14 (30.43%) patients of secondary wAIHA group, which is statistically insignificant. Venous thrombosis was observed in four (13.79%) patients of primary wAIHA group and 11 (23.91%) patients of secondary wAIHA group, which is statistically insignificant. Deaths were observed in three (10.34%) patients of primary wAIHA group and four (8.69%) patients of secondary wAIHA group, which is statistically insignificant.
WAIHA secondary to lymphoma
In our study, females constituted 11 and 27 patients of wAIHA secondary to lymphoma and other forms, while males constituted seven and 30 patients, respectively (P > 0.05). The common clinical characteristics of wAIHA secondary to lymphoma and other forms are represented in Table 3. Mean age of wAIHA onset was 69.7 ± 21.5 years in wAIHA group secondary to lymphoma and 54.3 ± 25.7 years in other wAIHA group, which is statistically significant. Regarding clinical symptoms at the time of onset, 14 (77.77%) patients in wAIHA group secondary to lymphoma and 45 (77.58%) patients in other wAIHA group reported it. Mean Hb level at AIHA onset was observed to be 6.4 ± 1.5 g/dL in wAIHA group secondary to lymphoma and 6.6 ± 1.8 g/dL in other wAIHA group, which is statistically not significant. Hypogammaglobulinemia was observed in 10 (55.55%) patients of wAIHA group secondary to lymphoma and six (10.34%) patients of other wAIHA group, which is statistically significant (P = 0.01). Monoclonal gammaglobulin was observed in 13 (72.22%) patients of wAIHA group secondary to lymphoma and 10 (17.24%) patients of other wAIHA group, which is statistically significant (P = 0.0019).
Click to view | Table 3. WAIHA Characteristics Secondary to Lymphoma |
Regarding DAT, IgG was observed in two (11.11%) patients of wAIHA group secondary to lymphoma and 30 (51.72%) patients of other wAIHA group, which is statistically significant (P = 0.029). IgG antibodies and complement (C3d) were observed in 14 (77.77%) patients of wAIHA group secondary to lymphoma and 28 (48.27%) patients of other wAIHA group, which is statistically insignificant. Only complement (C3d) was observed in 9% patients of wAIHA group secondary to lymphoma and 0% patients of other wAIHA group, which is statistically insignificant. IgA was observed in 0% patients of wAIHA group secondary to lymphoma and 4% patients of other wAIHA group, which is statistically insignificant.
Regarding treatment administered, blood transfusion was administered in 95% patients of wAIHA group secondary to lymphoma and 49% patients of other wAIHA group, which is statistically significant. Response to corticosteroids was observed in 94% patients of wAIHA group secondary to lymphoma and 90% patients of other wAIHA group, which is statistically insignificant. Dependence on corticosteroids was observed in 94% patients of wAIHA group secondary to lymphoma and 58% patients of other wAIHA group, which is statistically significant. Second line of treatment was administered in 96% patients of wAIHA group secondary to lymphoma and 51% patients of other wAIHA group, which is statistically significant. Splenectomy was done in 36% patients of wAIHA group secondary to lymphoma and 11% patients of other wAIHA group, which is statistically significant.
The most common cases of secondary wAIHA are represented in Table 4. They are B-cell lymphoma, SLE, rheumatoid arthritis, CLL, common variable immune deficiency (CVID), renal cell carcinoma and secondary to drug usage (alpha methyldopa and carbamazepine).
Click to view | Table 4. Most Common Causes of Secondary wAIHA |
| Discussion | ▴Top |
WAIHA is one of four clinical types of AIHA, characterized by the autoantibodies directing against patient’s own antigens on RBCs with the best reactive temperatures at 37 °C and accounting for 50-70% of all AIHA patients [43]. It produces a variable anemia, i.e., mild and sometimes severe. The secondary conditions causing wAIHA are primary immunodeficiencies such as common variable immunodeficiency, infections, hematologic malignancies, tumors, or drugs. Primary WAIHA comprises about 50% of patients.
CLL and lymphomas account for about half of secondary WAIHA cases. Autoimmune diseases, particularly SLE, account for a considerable proportion of secondary WAIHA cases. IgA class antibodies are present in about 14% of patients with WAIHA and almost always occur associated with IgG or IgM [44, 45]. Immunotherapy is defined as “the treatment of disease by inducing, enhancing, or suppressing an immune response”. It is classified as activation immunotherapy to elicit or amplify an immune response and suppression immunotherapy to reduce or suppress the immune response. The mainstay of therapy of wAIHA is immunosuppression with corticosteroids.
Allgood and Chaplin observed 43 patients, of whom 32 (74%) patients responded to corticosteroids but 25 (78%) patients of the responders group relapsed in the 9 months to 11 years of follow-up [46]. Zupanska et al demonstrated similar results with the most of patients responding initially, but only 19 (46%) patients of the 41 continued to respond after 3 weeks to treatment [47]. These studies suggested that 80% of patients respond to corticosteroids promptly; however, a proportion of responders relapse after the steroid-induced remission. Petz observed that about 50% of responders to corticosteroid require maintenance therapy [48]. For non-responders, usage of cyclosporin was documented by Pogglitsch et al [49].
Rituximab is a humanized monoclonal antibody directed against CD20 on pre-B cells and mature B lymphocytes. Binding of rituximab to CD20-positive cells causes cell death through a combination of antibody-dependent cell cytotoxicity, complement activation, and apoptosis [50, 51]. Quartier et al used rituximab for six children with refractory wAIHA and achieved a 100% CR rate [52]. Zecca et al treated 13 refractory wAIHA pediatric patients and observed a CR for 11, while two failed to respond [53].
In the adults, D’Arena et al treated 11 patients with refractory primary wAIHA by rituximab with a standard dose of 375 mg/m2 in a retrospective study [54]. Eight patients were observed to have a CR, three patients achieved a PR, but six patients still had laboratory signs of hemolysis. All patients remained in either CR or PR, at a mean follow-up of 604 days. Several retrospective studies in population of refractory primary or secondary AIHA confirmed the efficacy of rituximab [54-59]. The potential long-term complications of rituximab were reported by Carson et al [60], i.e., progressive multifocal leukoencephalopathy in two patients in 57 cases.
Barcellini et al [61] performed a clinical trial of low-dose rituximab (100 mg fixed dose for four weekly infusions) to investigate the efficacy, safety, and duration of response along with a short course of steroids as first- or second-line therapy in 23 patients with primary AIHA. They showed 82.6% overall response at month +2, improving to 90% at months +6 and +12, and the better response in wAIHA (100% overall response) than in cold AIHA (average, 60%). Several studies showed rituximab usage in refractory SLE-associated wAIHA with adequate safety and efficacy [62-64].
In an Exploratory Phase II/III SLE Evaluation of Rituximab (EXPLORER), the double-blind trial demonstrated that rituximab decreased B cells and anti-dsDNA autoantibody with increased C3 and C4 levels [65]. In a retrospective study of 53 patients with refractory AIHA administered rituximab in Belgium, the overall response rate was 79% with 47% CR and 32% of PR [56]. Mild infusion reaction including hypotension and fever is the most common complication observed with rituximab, and incidence of serious infection is very low [66]. Another recently developed monoclonal antibody, alemtuzumab, might be useful in treating wAIHA. Alemtuzumab is a humanized IgG1-type mAb directed against CD52 expressed on human B and T cells, natural killer cells, eosinophils, and macrophages [67, 68]. Alemtuzumab kills target cells by complement- and/or antibody-dependent cellular cytotoxicity, but also capable of inducing direct apoptosis via caspase-dependent and -independent mechanisms [69, 70].
Conclusion
We put forward the following observations based on our retrospective study. No significant difference between males and females was observed. Mean Hb level at AIHA onset was significantly lower in secondary wAIHA group. DAT pattern was similar in primary and secondary AIHA groups. There are no significant differences in occurrence of clinical features in primary and secondary AIHA groups. Splenectomies were more commonly done in secondary AIHA group compared to primary AIHA. Administration of corticosteroids, second-line treatments, pattern of remissions and complications were comparable without any statistically significant difference.
Significant difference in mean age of onset was observed in wAIHA secondary to lymphoma compared to others. Clinical features were comparable. Significant differences were observed in hypogammaglobulinemia, monoclonal gammaglobulin and IgG. Requirements of blood transfusion, dependence on corticosteroids, second line of treatment, and splenectomy were significantly higher in patients with wAIHA secondary to lymphoma. Our study shows effectiveness of corticosteroids as primary treatment and also efficacy and safety rituximab as second line of treatment. Splenectomy is useful in wAIHA secondary to lymphoma and should be considered whenever possible. Reducing the cumulative dose of corticosteroids with second-line treatment whenever possible and therefore reducing the risk of sepsis, specifically in older patients with comorbidities will reduce morbidity and mortality.
Abbreviations
CR: complete response; PR: partial response; AIHA: autoimmune hemolytic anemia; ACR: American College of Rheumatology; NHL: non-Hodgkin lymphoma; SLE: systemic lupus erythematosus; CVID: common variable immune deficiency; ALPD: autoimmune lymphoproliferative disease; SCT: stem cell transplantation
| References | ▴Top |
- Gallagher PG. The Red Blood Cell Membrane and Its Disorders: Hereditary Spherocytosis, Elliptocytosis, and Related Diseases. In: Kaushansky K, Lichtman MA, Beutler E, Kipps TJ, Seligsohn U, Prchal JT, eds. Williams Hematology. 8th ed. New York, NY: McGraw Hill; 2010. p. 617-646.
- Lichtman MA. Hemolytic anemia due to infections with microorganisms. In: Kaushansky K, Lichtman MA, Beutler E, Kipps TJ, Seligsohn U, Prchal JT, eds. Williams Hematology. 8th ed. New York, NY: McGraw Hill; 2010. p. 769-776.
- Beutler E, Bull BS, Herrmann PC. Hemolytic Anemia Resulting from Chemical and Physical Agents. In: Kaushansky K, Lichtman MA, Beutler E, Kipps TJ, Seligsohn U, Prchal JT, eds. Williams Hematology. 8th ed. New York, NY: McGraw Hill; 2010. p. 763-768.
- Glader BE. Hemolytic anemia in children. Clin Lab Med. 1999;19(1):87-111, vi.
pubmed - Kong JT, Schmiesing C. Concealed mothball abuse prior to anesthesia: mothballs, inhalants, and their management. Acta Anaesthesiol Scand. 2005;49(1):113-116.
doi pubmed - Lane DR, Youse JS. Coombs-positive hemolytic anemia secondary to brown recluse spider bite: a review of the literature and discussion of treatment. Cutis. 2004;74(6):341-347.
pubmed - Packman CH, Leddy JP. Acquired hemolytic anemia due to warm-reacting autoantibodies. In: Beutler E, Lichtman MA, Coller BS, Kipps TJ, eds. Williams Hematology. 5th ed. New York, NY: McGraw Hill; 1995. p. 667-684.
- Gallagher PG. Red cell membrane disorders. In: Hoffman R, Benz EJ Jr, Silberstein LE, Heslop H, Weitz J, Anastasi J, eds. Hematology: Basic Principles and Practice. 6th ed. New York, NY: Churchill Livingstone; 2013. p. 592-613.
- Price EA, Schrier SL. Extrinsic nonimmune hemolytic anemias. In: Hoffman R, Benz EJ Jr, Silberstein LE, Heslop H, Weitz J, Anastasi J, eds. Hematology: Basic Principles and Practice. 6th ed. New York, NY: Churchill Livingstone; 2013. p. 628-638.
- Jager U, Lechner K. Autoimmune hemolytic anemia. In: Hoffman R, Benz EJ Jr, Silberstein LE, Heslop H, Weitz J, Anastasi J, eds. Hematology: Basic Principles and Practice. 6th ed. New York, NY: Churchill Livingstone; 2013. p. 614-617.
- Shah A. Acquired hemolytic anemia. Indian J Med Sci. 2004;58(12):533-536.
pubmed - Berentsen S, Randen U, Tjonnfjord GE. Cold agglutinin-mediated autoimmune hemolytic anemia. Hematol Oncol Clin North Am. 2015;29(3):455-471.
doi pubmed - Naik R. Warm autoimmune hemolytic anemia. Hematol Oncol Clin North Am. 2015;29(3):445-453.
doi pubmed - Mayer B, Yurek S, Kiesewetter H, Salama A. Mixed-type autoimmune hemolytic anemia: differential diagnosis and a critical review of reported cases. Transfusion. 2008;48(10):2229-2234.
doi pubmed - Barros MM, Blajchman MA, Bordin JO. Warm autoimmune hemolytic anemia: recent progress in understanding the immunobiology and the treatment. Transfus Med Rev. 2010;24(3):195-210.
doi pubmed - Eaton WW, Rose NR, Kalaydjian A, Pedersen MG, Mortensen PB. Epidemiology of autoimmune diseases in Denmark. J Autoimmun. 2007;29(1):1-9.
doi pubmed - Cotran Ramzi S, Kumar Vinay, Fausto Nelson, Nelso Fausto, Robbins Stanley L, Abbas Abul K. Robbins and Cotran pathologic basis of disease. St. Louis, Mo: Elsevier Saunders. 2005; p. 637
- Gehrs BC, Friedberg RC. Autoimmune haemolytic anemia. Am J Hematol. 2002;69:258-271.
doi pubmed - Petz LD, Garraty G. Immune Haemolytic Anemias, 2nd Ed. Philadelphia, PA: Churchill Livingstone; 2004.
- Michel M. Classification and therapeutic approaches in autoimmune hemolytic anemia: an update. Expert Rev Hematol. 2011;4(6):607-618.
doi pubmed - Packman CH. Hemolytic anemia due to warm autoantibodies. Blood Rev. 2008;22(1):17-31.
doi pubmed - Genty I, Michel M, Hermine O, Schaeffer A, Godeau B, Rochant H. [Characteristics of autoimmune hemolytic anemia in adults: retrospective analysis of 83 cases]. Rev Med Interne. 2002;23(11):901-909.
doi - Valent P, Lechner K. Diagnosis and treatment of autoimmune haemolytic anaemias in adults: a clinical review. Wien Klin Wochenschr. 2008;120(5-6):136-151.
doi pubmed - Mauro FR, Foa R, Cerretti R, Giannarelli D, Coluzzi S, Mandelli F, Girelli G. Autoimmune hemolytic anemia in chronic lymphocytic leukemia: clinical, therapeutic, and prognostic features. Blood. 2000;95(9):2786-2792.
pubmed - Zent CS, Ding W, Reinalda MS, Schwager SM, Hoyer JD, Bowen DA, Jelinek DF, et al. Autoimmune cytopenia in chronic lymphocytic leukemia/small lymphocytic lymphoma: changes in clinical presentation and prognosis. Leuk Lymphoma. 2009;50(8):1261-1268.
doi pubmed - Gronbaek K, D'Amore F, Schmidt K. Autoimmune phenomena in non-Hodgkin's lymphoma. Leuk Lymphoma. 1995;18(3-4):311-316.
doi pubmed - Gertz MA. Cold hemolytic syndrome. Hematology Am Soc Hematol Educ Program. 2006:19-23.
doi pubmed - Lechner K, Chen YA. Paraneoplastic autoimmune cytopenias in Hodgkin lymphoma. Leuk Lymphoma. 2010;51(3):469-474.
doi pubmed - Puthenparambil J, Lechner K, Kornek G. Autoimmune hemolytic anemia as a paraneoplastic phenomenon in solid tumors: A critical analysis of 52 cases reported in the literature. Wien Klin Wochenschr. 2010;122(7-8):229-236.
doi pubmed - Payne D, Muss HB, Homesley HD, Jobson VW, Baird FG. Autoimmune hemolytic anemia and ovarian dermoid cysts: case report and review of the literature. Cancer. 1981;48(3):721-724.
doi - Jeffries M, Hamadeh F, Aberle T, Glenn S, Kamen DL, Kelly JA, Reichlin M, et al. Haemolytic anaemia in a multi-ethnic cohort of lupus patients: a clinical and serological perspective. Lupus. 2008;17(8):739-743.
doi pubmed - Giannadaki E, Potamianos S, Roussomoustakaki M, Kyriakou D, Fragkiadakis N, Manousos ON. Autoimmune hemolytic anemia and positive Coombs test associated with ulcerative colitis. Am J Gastroenterol. 1997;92(10):1872-1874.
pubmed - Seve P, Bourdillon L, Sarrot-Reynauld F, Ruivard M, Jaussaud R, Bouhour D, Bonotte B, et al. Autoimmune hemolytic anemia and common variable immunodeficiency: a case-control study of 18 patients. Medicine (Baltimore). 2008;87(3):177-184.
doi pubmed - Straus SE, Sneller M, Lenardo MJ, Puck JM, Strober W. An inherited disorder of lymphocyte apoptosis: the autoimmune lymphoproliferative syndrome. Ann Intern Med. 1999;130(7):591-601.
doi pubmed - Sanz J, Arriaga F, Montesinos P, Orti G, Lorenzo I, Cantero S, Puig N, et al. Autoimmune hemolytic anemia following allogeneic hematopoietic stem cell transplantation in adult patients. Bone Marrow Transplant. 2007;39(9):555-561.
doi pubmed - Elimelakh M, Dayton V, Park KS, Gruessner AC, Sutherland D, Howe RB, Reding MT, et al. Red cell aplasia and autoimmune hemolytic anemia following immunosuppression with alemtuzumab, mycophenolate, and daclizumab in pancreas transplant recipients. Haematologica. 2007;92(8):1029-1036.
doi pubmed - Dearden C. Disease-specific complications of chronic lymphocytic leukemia. Hematology Am Soc Hematol Educ Program. 2008:450-456.
doi pubmed - Chiao EY, Engels EA, Kramer JR, Pietz K, Henderson L, Giordano TP, Landgren O. Risk of immune thrombocytopenic purpura and autoimmune hemolytic anemia among 120 908 US veterans with hepatitis C virus infection. Arch Intern Med. 2009;169(4):357-363.
doi pubmed - Murphy S, LoBuglio AF. Drug therapy of autoimmune hemolytic anemia. Semin Hematol. 1976;13(4):323-334.
pubmed - Michel M, Chanet V, Dechartres A, Morin AS, Piette JC, Cirasino L, Emilia G, et al. The spectrum of Evans syndrome in adults: new insight into the disease based on the analysis of 68 cases. Blood. 2009;114(15):3167-3172.
doi pubmed - Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R, Derksen RH, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006;4(2):295-306.
doi pubmed - Vitali C, Bombardieri S, Moutsopoulos HM, Balestrieri G, Bencivelli W, Bernstein RM, Bjerrum KB, et al. Preliminary criteria for the classification of Sjogren's syndrome. Results of a prospective concerted action supported by the European Community. Arthritis Rheum. 1993;36(3):340-347.
doi pubmed - Stahl D, Sibrowski W. Warm autoimmune hemolytic anemia is an IgM-IgG immune complex disease. J Autoimmun. 2005;25(4):272-282.
doi pubmed - Sokol RJ, Booker DJ, Stamps R, Booth JR, Hook V. IgA red cell autoantibodies and autoimmune hemolysis. Transfusion. 1997;37(2):175-181.
doi pubmed - Meyer F, Garin L, Smati C, Gaspard M, Giannoli C, Rigal D. [Application of the gel test using and anti-IgA antiglobulin for the immunologic diagnosis of autoimmune hemolytic anemia with a negative direct Coombs test]. Transfus Clin Biol. 1999;6(4):221-226.
doi - Allgood JW, Chaplin H, Jr. Idiopathic acquired autoimmune hemolytic anemia. A review of forty-seven cases treated from 1955 through 1965. Am J Med. 1967;43(2):254-273.
doi - Zupanska B, Sylwestrowicz T, Pawelski S. The results of prolonged treatment of autoimmune haemolytic anaemia. Haematologia (Budap). 1981;14(4):425-433.
- Petz LD. Treatment of autoimmune hemolytic anemias. Curr Opin Hematol. 2001;8(6):411-416.
doi pubmed - Pogglitsch H, Warnkross H, Weinrauch V, Winkler HM. [Successful treatment of autoimmune hemolytic anemia with cyclosporin A]. Wien Med Wochenschr. 1986;136(21-22):567-570.
pubmed - Reff ME, Carner K, Chambers KS, Chinn PC, Leonard JE, Raab R, Newman RA, et al. Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood. 1994;83(2):435-445.
pubmed - Shan D, Ledbetter JA, Press OW. Apoptosis of malignant human B cells by ligation of CD20 with monoclonal antibodies. Blood. 1998;91(5):1644-1652.
pubmed - Quartier P, Brethon B, Philippet P, Landman-Parker J, Le Deist F, Fischer A. Treatment of childhood autoimmune haemolytic anaemia with rituximab. Lancet. 2001;358(9292):1511-1513.
doi - Zecca M, Nobili B, Ramenghi U, Perrotta S, Amendola G, Rosito P, Jankovic M, et al. Rituximab for the treatment of refractory autoimmune hemolytic anemia in children. Blood. 2003;101(10):3857-3861.
doi pubmed - D'Arena G, Califano C, Annunziata M, Tartarone A, Capalbo S, Villani O, Amendola G, et al. Rituximab for warm-type idiopathic autoimmune hemolytic anemia: a retrospective study of 11 adult patients. Eur J Haematol. 2007;79(1):53-58.
doi pubmed - Bussone G, Ribeiro E, Dechartres A, Viallard JF, Bonnotte B, Fain O, Godeau B, et al. Efficacy and safety of rituximab in adults' warm antibody autoimmune haemolytic anemia: retrospective analysis of 27 cases. Am J Hematol. 2009;84(3):153-157.
doi pubmed - Dierickx D, Verhoef G, Van Hoof A, Mineur P, Roest A, Triffet A, Kentos A, et al. Rituximab in auto-immune haemolytic anaemia and immune thrombocytopenic purpura: a Belgian retrospective multicentric study. J Intern Med. 2009;266(5):484-491.
doi pubmed - Narat S, Gandla J, Hoffbrand AV, Hughes RG, Mehta AB. Rituximab in the treatment of refractory autoimmune cytopenias in adults. Haematologica. 2005;90(9):1273-1274.
pubmed - Shanafelt TD, Madueme HL, Wolf RC, Tefferi A. Rituximab for immune cytopenia in adults: idiopathic thrombocytopenic purpura, autoimmune hemolytic anemia, and Evans syndrome. Mayo Clin Proc. 2003;78(11):1340-1346.
doi pubmed - Zaja F, Iacona I, Masolini P, Russo D, Sperotto A, Prosdocimo S, Patriarca F, et al. B-cell depletion with rituximab as treatment for immune hemolytic anemia and chronic thrombocytopenia. Haematologica. 2002;87(2):189-195.
pubmed - Carson KR, Evens AM, Richey EA, Habermann TM, Focosi D, Seymour JF, Laubach J, et al. Progressive multifocal leukoencephalopathy after rituximab therapy in HIV-negative patients: a report of 57 cases from the Research on Adverse Drug Events and Reports project. Blood. 2009;113(20):4834-4840.
doi pubmed - Barcellini W, Zaja F, Zaninoni A, Imperiali FG, Battista ML, Di Bona E, Fattizzo B, et al. Low-dose rituximab in adult patients with idiopathic autoimmune hemolytic anemia: clinical efficacy and biologic studies. Blood. 2012;119(16):3691-3697.
doi pubmed - Conti F, Perricone C, Ceccarelli F, Valesini G. Rituximab treatment of systemic lupus erythematosus in controlled trials and in clinical practice: Two sides of the same coin. Autoimmun Rev. 2010;9(11):716-720.
doi pubmed - Tanaka Y, Yamamoto K, Takeuchi T, Nishimoto N, Miyasaka N, Sumida T, Shima Y, et al. A multicenter phase I/II trial of rituximab for refractory systemic lupus erythematosus. Mod Rheumatol. 2007;17(3):191-197.
doi pubmed - Reynolds JA, Toescu V, Yee CS, Prabu A, Situnayake D, Gordon C. Effects of rituximab on resistant SLE disease including lung involvement. Lupus. 2009;18(1):67-73.
doi pubmed - Merrill J, Buyon J, Furie R, Latinis K, Gordon C, Hsieh HJ, Brunetta P. Assessment of flares in lupus patients enrolled in a phase II/III study of rituximab (EXPLORER). Lupus. 2011;20(7):709-716.
doi pubmed - Newman K, Owlia MB, El-Hemaidi I, Akhtari M. Management of immune cytopenias in patients with systemic lupus erythematosus - Old and new. Autoimmun Rev. 2013;12(7):784-791.
doi pubmed - Hale G, Xia MQ, Tighe HP, Dyer MJ, Waldmann H. The CAMPATH-1 antigen (CDw52). Tissue Antigens. 1990;35(3):118-127.
doi pubmed - Treumann A, Lifely MR, Schneider P, Ferguson MA. Primary structure of CD52. J Biol Chem. 1995;270(11):6088-6099.
doi pubmed - Mone AP, Cheney C, Banks AL, Tridandapani S, Mehter N, Guster S, Lin T, et al. Alemtuzumab induces caspase-independent cell death in human chronic lymphocytic leukemia cells through a lipid raft-dependent mechanism. Leukemia. 2006;20(2):272-279.
doi pubmed - Stanglmaier M, Reis S, Hallek M. Rituximab and alemtuzumab induce a nonclassic, caspase-independent apoptotic pathway in B-lymphoid cell lines and in chronic lymphocytic leukemia cells. Ann Hematol. 2004;83(10):634-645.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.