

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website http://www.thejh.org |
Case Report
Volume 6, Number 1, March 2017, pages 25-28
Transient Abnormal Myelopoiesis: A Varied Spectrum of Clinical Presentation
Amitabh Singha, Anirban Mandalb, Vijay Guruc, Sindhu Srinivasand, Rachna Sethc, e
aDepartment of Pediatrics, Chacha Nehru Bal Chikitsalaya, New Delhi, India
bDepartment of Pediatrics, Sitaram Bhartia Institute of Science and Research, New Delhi, India
cDivision of Pediatric Oncology, Department of Pediatrics, All India Institute of Medical Sciences, New Delhi, India
dDivision of Neonatology, Department of Pediatrics, All India Institute of Medical Sciences, New Delhi, India
eCorresponding Author: Rachna Seth, Division of Pediatric Oncology, Department of Pediatrics, All India Institute of Medical Sciences, Ansari Nagar, New Delhi 110029, India
Manuscript accepted for publication December 16, 2016
Short title: Transient Abnormal Myelopoiesis
doi: https://doi.org/10.14740/jh306w
| Abstract | ▴Top |
Transient myeloproliferative disorder (TMD) is a condition seen almost exclusively in newborns with Down syndrome (DS). It can have a spectrum of clinical presentation ranging from being asymptomatic with incidental detection to a stormy course and fatal outcome. We describe three cases of TMD having different clinical presentation, course, complications and outcome. All but one had Down’s phenotype; one of them had a severe disease with tumor lysis syndrome and died of liver failure, while the other one had pericardial effusion and cardiac failure but survived. The third patient had a very benign course of illness requiring only supportive care. Newborns with DS should be screened for TMD by a complete blood count during their first month of life, irrespective of symptoms. With increasing knowledge about the natural history and management guidelines, the prognosis of this rare and unique entity has improved in recent years.
Keywords: Transient myelopoiesis; Tumor lysis syndrome; Down syndrome; Hyperleukocytosis
| Introduction | ▴Top |
Transient myeloproliferative disorder (TMD) or transient abnormal myelopoiesis (TAM) is described almost exclusively in newborns with Down syndrome (DS). About 4-10% of newborns with DS develop TMD [1]. Majority of such children have a milder clinical course, requiring only supportive measures; there is gradual decrease in blasts followed by spontaneous remission within the first 3 months in almost 80% of cases. However, they remain at a high risk of developing non-transient acute megakaryoblastic leukemia (AMKL), occurring in around 20-30% of all cases. In a minority of cases, TMD presents with an aggressive course and fatal outcome [2]. Very rarely, TMD has also been described in newborn without DS, although, almost always in association with trisomy 21 and/or GATA1 mutation [3].
Here we describe three cases of TAM having different clinical presentations, illustrating the widely variable spectrum of this clinical entity.
| Case Reports | ▴Top |
Case 1
A male baby was born by normal vaginal delivery (NVD) to a 35-year-old primigravida, at 37 weeks of gestation with a birth weight of 2.7 kg. Antenatal and perinatal periods were uneventful. He had Down’s phenotype (mongoloid slant of eyes, flat facial features, flat occiput, clinodactyly and antenatally diagnosed atrio-ventricular septal defect (AVSD)) with hepatosplenomegaly. At 48 h of life, baby developed deceasing urine output with deranged renal function test (RFT) (urea 103 mg/dL and creatinine 2.8 mg/dL); serum biochemistry revealed calcium-phosphate product of 81 with very high uric acid (14 mg/dL) but serum electrolytes were normal. Total leucocyte count (TLC) was 158,000/mm3, with peripheral smear showing 18% blasts; flow cytometry further confirmed that the blasts were of myeloid origin (CD34+, CD 45+, CD11b+, CD56+, CD33+, CD38+, CD117+, HLA-DR+, CD9+, CD13-, CD61-, CD19-, CD10-, CD14-, CD64-, CD15-, CD2-, CD65-, CD3-, CD79a-, and CMPO-). Baby was diagnosed with tumor lysis syndrome (TLS) with acute kidney injury (AKI) secondary to TAM. GATA1 mutation could not be done due to non-availability. He was started on controlled hydration, diuretics and allopurinol along with strict monitoring. Blast percentage showed a decrease and clinical and laboratory features of TLS also settled over next 5 days. There was also liver dysfunction with conjugated hyperbilirubinemia (total serum bilirubin 13.5 mg/dL and conjugated bilirubin 6.2 mg/dL) along with deranged liver function test (LFT) and prothrombin time (PT) > 1 min from day 3 of life. As common causes of neonatal cholestasis (e.g. congenital infections, hypothyroidism, inborn error of metabolism, billiary atresia, etc.) were ruled out with appropriate investigations, it was attributed to TMA. Cholestasis regimen was started but deepening jaundice along with progressive increase in liver dysfunction, though the baby remained clinically stable. Karyopyting was suggestive of trisomy 21 and the parents did not agree for chemotherapy. He required multiple blood product therapy but there was no clinically apparent bleeding. On day 47 of life, baby developed worsening respiratory distress and edema and expired within next 2 days. Post mortem liver biopsy showed extensive intracellular cholestasis, feathery degeneration, giant cell transformation and acinar formation with extensive iron deposition, features suggestive of obstructive cholangiopathy with diffuse hepatic fibrosis and secondary hemosiderosis.
Case 2
A low birth weight (1.9 kg), preterm (34 weeks), male baby was born by NVD to a 33-year-old, sixth gravida mother with an uneventful antenatal period. Baby did not require any resuscitation, but had respiratory distress at birth requiring oxygen inhalation. Investigations done in view of persistent tachypnea revealed a TLC of 140,000/mm3 with abnormal cells in peripheral smear on day 4 of life. Workup for sepsis was negative, and the baby was referred and presented to us on day 6 of life. At presentation, he had tachypnea with mild chest retractions but was normoxemic and hemodynamically stable. There was Down’s phenotype (mongoloid slant of eyes, flat facial features, flat occiput, sandle gap and hypotonia), icterus till thigh, hepatosplenomegaly with an ejection systolic murmur grade 3/6. The TLC was 138,000/mm3 with peripheral smear showing 89% MPO negative blasts (Fig. 1a). Peripheral blood flow cytometry revealed blasts with CD34+, CD45+, CD13+, CD33+, 30% CD61, 41(+), suggestive of TMD. GATA1 gene mutation could not be done. Chest X-ray had cardiomegaly (Fig. 1b); echocardiography confirmed presence of atrial septal defect (ASD) and ventricular septal defect (VSD) with pericardial effusion. Biochemical investigations (LFT, RFT and serum electrolytes) were within normal limits. Consultation with pediatric cardiology was done and the baby was treated conservatively (oxygen inhalation, careful hydration, diuretics, allopurinol, and hydroxyurea) with intensive clinical and laboratory monitoring. The jaundice was physiological, requiring no intervention. The TLC gradually decreased to normal and respiratory distress settled over next 7 days. Karyotyping confirmed DS. The baby was discharged after 10 days. By 6 weeks, TLC was within normal range for age with no detectable blasts in peripheral smear. The pericardial effusion subsided after 1 month. Last bone marrow examination at 3 months also did not reveal any increase in blasts. On follow-up for 6 months, he remained asymptomatic except failure to thrive (Fig. 1c) and is scheduled to undergo cardiac surgery after appropriate weight gain.
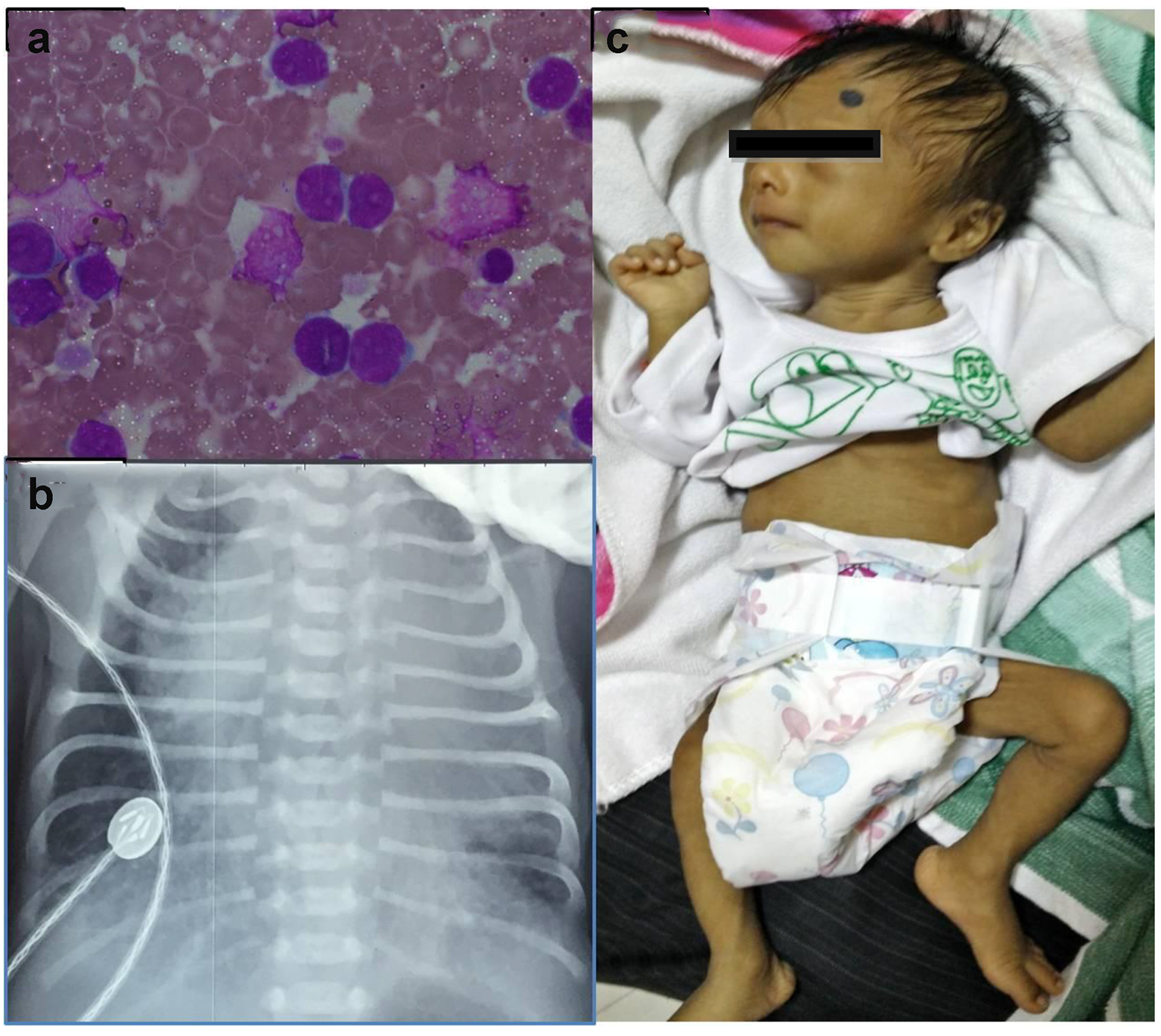 Click for large image | Figure 1. (a) Peripheral smear showing blast. (b) Chest X-ray showing cardiomegaly with increased pulmonary blood flow. (c) The baby after 6 months of follow-up. |
Case 3
A 3.1 kg healthy, female newborn was born to a 29-year-old primigravida at term by NVD. There were no antenatal or perinatal concerns. Routine examination at 24 h of life revealed hepatosplenomegaly and subsequently done investigations were suggestive of leucocytosis (TLC 38,000/mm3) with thrombocytopenia (platelet count 52,000/mm3) but no anemia (hemoglobin 15.1 g/dL). Peripheral smear showed 35% blasts which were CD34+, CD45+, CD11b+, CD56+, CD33+, CD38+, CD117+ and HLA-DR+. Serum biochemistry was within normal limits except an elevated uric acid level of 10.2 mg/dL. There was no characteristic phenotypic features of Down’s phenotype and workup for other common causes of a leukemoid reaction (e.g. sepsis, congenital infections, etc.) was negative. Bone marrow examination was done on day 4 of life; there was 28% blasts with trisomy 21 and no other cytogenetic abnormalities. In the absence of GATA1 mutation analysis facilities, a clinical diagnosis of TMD was made and the child was started on hydration (oral and intravenous) and allopurinol along with strict clinical and laboratory monitoring for TLS. Apart from exaggerated physiological hyperbilirubinemia requiring 24 h of phototherapy and mildly elevated liver enzymes, the baby remained well. In follow-up, the TLC normalized at 3 months with no evidence of blasts in peripheral blood since 2 months. After 1 year of follow-up, the child remains asymptomatic and well thriving.
| Discussion | ▴Top |
The true incidence of TAM is unknown, as many (10-25%) remain asymptomatic and routine laboratory screening of DS newborns are not being done. Most commonly, it presents between 3 and 7 days, but may be delayed till 2 months. Commonest clinical manifestations include hepatomegaly (60%), splenomegaly (35-40%), jaundice (15%), pericardial effusion (15%), pleural effusion (10-15%), ascites (10%), respiratory distress (10%), and bleeding diathesis (10%), while less common features include hepatic fibrosis, hydrops fetalis, and renal failure [4]. The child in case 1 had hepatosplenomegaly, jaundice with hepatic fibrosis and renal failure; case 2 had pericardial effusion in addition to hepatosplenomegaly while the third case presented only with incidentally detected organomegaly. American Academy of Pediatrics (AAP), therefore, recommends screening of all DS newborns for TMD by obtaining a complete blood count once in the first month of life, irrespective of symptoms [5].
The diagnosis of TMD is made in a newborn with DS with leukocytosis, blasts in peripheral blood with or without thrombocytopenia and organomegaly [4]. The blasts in TMD generally express stem cell (CD34 and CD117), myeloid (CD13 and CD33), no lineage (CD4, CD7, and CD56), and megakaryoblastic (CD61, CD41, and CD42) antigens [6]. In the first two cases, the diagnosis was made with the help of a combination of clinical (DS with organomegaly) and pathological (peripheral smear and peripheral blood flow cytometry) features but in the third case it was the bone marrow cytogenetics showing trisomy 21 that clinched the diagnosis.
The differential diagnosis of TAM includes leukemoid reactions, erythroblastosis fetalis, congenital infections and congenital leukemia [7]. No critical threshold for blast percentage for diagnosis of TAM is established, so it becomes very difficult to distinguish it from congenital leukemia. Younger age at presentation, higher hemoglobin concentrations and platelet counts and the unique finding of a higher percentage of blast cell in peripheral blood than in marrow point toward TAM, but it is the non-resolution by 3 months of age that really defines it to be a non-transient leukemia [1, 2].
Very few cases of TAM with TLS are reported in literature, either spontaneous or after chemotherapy and managed with hydration, diuretics and rasburicase [8]. Our first patient had spontaneous TLS and renal failure, managed successfully with careful hydration, diuretics and allopurinol. Presence of congenital heart disease made the scenario further challenging.
The newborn in first case died of liver failure secondary to hepatic fibrosis. Almost 20% cases of TAM develop cholestasis or elevation of liver enzymes [9]. Early (< 6 months) mortality in TMD is seen in approximately one-fifth of cases attributed to organ infiltration and failure, greatest risk being involved with hepatic failure [1, 2]. Fetal liver being the predominant origin of blasts, megakaryoblast infiltration and/or megakaryoblast-induced hepatic fibrosis mediated by platelet-derived growth factor (PDGF) and transforming growth factor beta 1 (TGFB1) may be responsible for the hepatic fibrosis seen in TAM [2]. The management of liver disease in TMD is largely supportive with adequate nutrition, fat soluble vitamin supplementation and ursodeoxycholic acid (UDCA) [9].
Management of TAM depends on its clinical manifestation with a goal of symptom management until natural resolution. Only careful monitoring suffices in the milder cases with supportive care being the cornerstone in most of the symptomatic patients. But in the presence of one or more life-threatening features, i.e., signs of hyperviscosity, blast count > 100,000/µL, hepatosplenomegaly causing respiratory compromise, heart failure not directly because of a congenital heart defect, hydrops fetalis, renal or hepatic dysfunction, and disseminated intravascular coagulation with bleeding, intervention is advised [1]. Low dose cytarabine arabinoside therapy in such cases has decreased mortality substantially. The largest study on TMD shows that about 22% of patients with severe TMD ultimately required intervention [1]. In presence of hepatic dysfunction though, the success rate of chemotherapy is significantly decreased along with increased chances of adverse effects [9].
With a case fatality rate of approximately 20%, only half of them were attributed to TMD itself [1]. The mortality in our series was also attributed to liver failure. The risk factors for mortality in TMD include hyperleucocytosis, hepatic dysfunction, effusions, prematurity, low birth weight, renal failure, etc. [2]. Although the newborn in second case had many of these risk factors, we could manage him successfully.
The biological mechanism of spontaneous resolution in TMS is unclear with diminished telomerase activity in the TMD blasts being one of the suggested explanations [10]. It is also not known whether the development of AMKL is recurrence of original disease or appearance of new disease. Thus, the 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia [11] classified these two entities under a common category termed “myeloid proliferations related to DS”. But prior to onset of AML, there is often a period of several months characterized by thrombocytopenia and bone marrow myelofibrosis with dysplastic megakaryocytes [7]. Unfortunately, there is no test till date which can predict the future development of AMKL, therefore, regular follow-up with blood counts every 3 - 6 months for the first 4 years of life along with education of the parents is advised [1, 2]. We are also following up the two children with transient disorder; even if they develop leukemia later in life, it is known to be very well responsive to therapy [2].
Conflicts of Interest
The authors declare that they have no conflicts of interest.
| References | ▴Top |
- Gamis AS, Alonzo TA, Gerbing RB, Hilden JM, Sorrell AD, Sharma M, Loew TW, et al. Natural history of transient myeloproliferative disorder clinically diagnosed in Down syndrome neonates: a report from the Children's Oncology Group Study A2971. Blood. 2011;118(26):6752-6759; quiz 6996.
doi pubmed - Gamis AS, Smith FO. Transient myeloproliferative disorder in children with Down syndrome: clarity to this enigmatic disorder. Br J Haematol. 2012;159(3):277-287.
doi pubmed - Rozen L, Huybrechts S, Dedeken L, Heijmans C, Dessars B, Heimann P, Lambert F, et al. Transient leukemia in a newborn without Down syndrome: case report and review of the literature. Eur J Pediatr. 2014;173(12):1643-1647.
doi pubmed - Bombery M, Vergilio JA. Transient abnormal myelopoiesis in neonates: GATA get the diagnosis. Arch Pathol Lab Med. 2014;138(10):1302-1306.
doi pubmed - Bull MJ. Health supervision for children with Down syndrome. Pediatrics. 2011;128(2):393-406.
doi pubmed - Langebrake C, Creutzig U, Reinhardt D. Immunophenotype of Down syndrome acute myeloid leukemia and transient myeloproliferative disease differs significantly from other diseases with morphologically identical or similar blasts. Klin Padiatr. 2005;217(3):126-134.
doi pubmed - Sajid N, Ahmed N, Mahmood S. Clinicopathological features of transient myeloproliferative syndrome and congenital leukaemia. J Coll Physicians Surg Pak. 2010;20(9):576-580.
pubmed - Tragiannidis A, Pana ZD, Papageorgiou T, Hatzipantelis E, Hatzistilianou M, Athanassiadou F. Transient myeloproliferative disorder in a newborn with down syndrome treated with rasburicase for the risk of development of tumor lysis syndrome: A case report. J Med Case Rep. 2011;5:407.
doi pubmed - Park MJ, Sotomatsu M, Ohki K, Arai K, Maruyama K, Kobayashi T, Nishi A, et al. Liver disease is frequently observed in Down syndrome patients with transient abnormal myelopoiesis. Int J Hematol. 2014;99(2):154-161.
doi pubmed - Holt SE, Brown EJ, Zipursky A. Telomerase and the benign and malignant megakaryoblastic leukemias of Down syndrome. J Pediatr Hematol Oncol. 2002;24(1):14-17.
doi pubmed - Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, Harris NL, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937-951.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.