

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website http://www.thejh.org |
Original Article
Volume 5, Number 4, December 2016, pages 123-128
Beta-Globin Haplotypes and Alpha-Thalassemia 3.7 kb Deletion in Sickle Cell Disease Patients From the Occidental Brazilian Amazon
Janaina Santana Carneiroa, Marilda de Souza Goncalvesb, Sergio Roberto Lopes Albuquerqueb, Nelson Abrahim Fraijib, Jose Pereira de Moura Netoa, c, d
aFundacao Hospitalar de Hematologia e Hemoterapia do Amazonas, Manaus, Amazonas, Brazil
bFundacao Oswaldo Cruz - Centro de Pesquisas Goncalo Moniz, Salvador, Bahia, Brazil
cUniversidade Federal do Amazonas, Faculdade de Ciencias Farmaceuticas, Manaus, Amazonas, Brazil
dCorresponding Author: Jose Pereira de Moura Neto, Universidade Federal do Amazonas, Faculdade de Ciencias Farmaceuticas, Avenida General Rodrigo Otavio Jordao Ramos, 6200 - Coroado I, Manaus, AM, 69067-005, Brazil
Manuscript accepted for publication December 27, 2016
Short title: β-Globin Haplotypes and α-Thalassemia in SCD
doi: https://doi.org/10.14740/jh310w
| Abstract | ▴Top |
Background: Sickle cell disease (SCD) includes a group of inherited red blood cell disorders. SCD patients vary widely from person to person. We describe βS and βC haplotypes and α-thalassemia 3.7 kb genotypes from SCD patients Fundacao Hospitalar de Hematologia e Hemoterapia do Amazonas, Manaus, AM.
Methods: Our survey included 139 HbSS and 11 HbSC patients. Molecular genotypes have been identified by PCR-RFLP and α-thalassemia 3.7 kb deletion by ASO-PCR. Male/female distribution was 42.3%/57.7%.
Results: The average age at enrolment was 19.1 years for HbSS and 25.85 years for HbSC. Average fetal hemoglobin was 11.27% for HbSS and 7.86% for HbSC. Anemia in HbSS patients was more severe and hemolysis twice as stronger as HbSC individuals. The frequency distribution of the most common β-globin haplotypes among HbSS patients was 52.5% CAR/CAR, 23.7% CAR/Ben and 18% Ben/Ben. For the HbSC group, the haplotype distribution was 36.3% CAR/CI, 27.3% Benin/CI, 18.2% CAR/CII, 9.1% CAR/CIII and 9.1% Benin/CII. Of the HbSS patients, 13.7% and 2.8% were heterozygous and homozygous for the α-thalassemia 3.7 kb deletion, respectively. No HbSC patients presented the deletion.
Conclusions: Here we present the distribution of haplotypes βS and βC and α-thalassemia deletion 3.7 kb in a population sample with SCD from the Occidental Brazilian Amazon. Results have been analyzed in the context of hematological and biochemical profiles.
Keywords: Sickle cell disease; α-thalassemia; Haplotypes; Amazon patients
| Introduction | ▴Top |
Sickle cell disease (SCD) is a severe genetic disorder that affects populations around the world and is characterized by the presence of hemoglobin S (HbS) inside the erythrocyte. The condition is caused by a point mutation GAG>GTG on the sixth position of the β-globin gene, resulting in the replacement of glutamic acid by valine in the polypeptide beta chain (βS) [1]. A variation GAG>AAG in the same locus induces a change from glutamic acid to lysine that characterizes the βC chain. Homozygosis for βS results in a disorder known as sickle cell anemia (HbSS); individual’s heterozygous βSβC presents hemoglobin SC (HbSC) disease [2, 3].
HbSS patients present atypical red blood cells (RBCs) with classic sickle-shape that do not circulate properly in the microcirculation, causing blood flow obstruction, hemolysis and vaso-occlusive crisis (VOC) [4]. These events are responsible for the main clinical manifestations of the disease, as well as for significant morbidity and reduction of life expectancy [5]. HbSC patients present target-shape RBC and manifest similar clinical features of those with HbSS, however, in a lower frequency and intensity [6, 7].
Hematologic and biochemical parameters, as well as clinical manifestation of both HbSS and HbSC diseases, are influenced by genetic factors such as haplotypes of the β-globin gene cluster and the α-thalassemia 3.7 kb deletion [8-11]. Different haplotypes linked to the βS globin gene have been described in SCD patients from Senegal (Sen), Benin (Ben), Central African Republic (CAR), Cameroon (Cam) and Arab-Indian (Arab) and have been named according to the geographical region or ethnic group in which they have been originally identified. Among HbSC patients, haplotype groups have been characterized as Sen/I, Sen/II, Sen/III, Ben/I, Ben/II, Ben/III, Car/I, Car/II, and Car/III [8, 9, 12].
The presence of α-thalassemia in patients with SCD is associated to milder phenotypes, as the polymerization potential of HbS and HbC decreases, leading to the presence of fewer dense cells with little deforming and increased hematocrit, reducing the vaso-occlusive events and preserving spleen function [6, 9, 13].
Although molecular characterization surveys of populations affected by SCD have been carried out in several countries including Brazil, results have been often conflicting [14-19]. In addition, for some particularly interesting regions of Brazil, such as the occidental Amazon, this molecular profile is still widely unknown. Finally, few studies have compared biochemical and hematological parameters across SCD individuals of different β-globin and α-thalassemia molecular backgrounds. Here we describe the β-globin and α-thalassemia genetic profile of a population sample of individuals affected by sickle cell and hemoglobin SC disease from the Brazilian Amazon, presented and discussed in the context of a comprehensive hematological and biochemical profile [20-23].
| Materials and Methods | ▴Top |
The studied population sample was composed by 150 SCD patients attending the Hematological Hospital of the Fundacao Hospitalar de Hematologia e Hemoterapia do Amazonas (FHEMOAM) located in the city of Manaus, capital of the state of Amazonas, Brazil. Of these, 139 were homozygotes HbSS and 11 were double heterozygous HbSC. Gender distribution was 87 (58%) females and 63 (42%) males, with age at enrolment between 4 months and 57 years old. Average age at diagnosis was 6.5 ± 11.23 years for HbSS and 10 ± 12.52 years for HbSC individuals. Hemoglobin profiles were confirmed by high-performance liquid chromatography (HPLC) (Bio-Rad, Hercules, CA, USA). All participants or guardians (in the case of children under 18 years of age) signed the written consent form and the study was approved by the Research Ethics Committee (CEP) of the Federal University of Amazonas (UFAM) under the CAAE number 37941514.4.0000.5020.
Peripheral blood samples for the hematological and biochemical analysis were obtained during a routine appointment for follow-up. The hematological analysis was performed using the automated hematologic analyzer BC-5800 (Mindray, Shenzhe, China); data were obtained for the overall count of RBCs, concentration of hemoglobin, hematocrit, mean corpuscular volume (MCV), mean corpuscular hemoglobin (MCH), mean corpuscular hemoglobin concentration (MCHC), red blood cell distribution width (RDW), total and differential leukocyte count and platelet count. Fetal hemoglobin (HbF) detection was performed using the alkaline resistance biochemical test [24]. Biochemical parameters were measured as implemented in the automated A25 platform (BioSystems SA, Barcelona, Spain) and included serum concentration of urea, creatinine, direct and indirect bilirubin, glucose, triglycerides, iron and ferritin, as well as gamma-glutamyl transferase (GGT) and lactate dehydrogenase activity (LDH).
Genomic DNA was obtained from peripheral blood using HiYield Genomic DNA extraction kit (BioAmerica Inc., USA). NanoDrop ND-1000 (Isogen Life Science, the Netherlands) was used to measure DNA concentration. Seven specific loci of the β-globin gene located in the short arm of chromosome 11 were amplified and genotyped by PCR-RFLP, as described previously [25]. In brief, the amplified fragments were digested by the following restriction enzymes: Xmn I (position 5' of the γG), Hinc II (pseudogene ψβ), Hinf I and Hpa I (of regions 5' and 3' of the β-gene, respectively). After digestion, DNA fragments were separated by electrophoresis in agarose gel in 1% under a constant 80 V for 45 min and visualized under ultraviolet light.
Genotyping of the α-thalassemia 3.7 kb deletion was performed by ASO-PCR as described by Baysal and Huisman, using the proposed nomenclature for normal (A + C) or mutated (A + B) genotypic profiles. The PCR products were submitted to electrophoresis (Bio-Rad, EUA) in agarose gel in 1% under a constant 80 V for 45 min and visualized under ultraviolet light. Patients were distributed into three groups: normal (A + C), heterozygous (A + C) + (A + B) and homozygous (A + B) for the deletion [26].
Statistical analyzes were performed using one-way ANOVA and Kruskal-Wallis test, as implemented in SPSS, version 22.0. P-values < 0.05 were considered significant.
| Results | ▴Top |
Table 1 summarizes and compares the hematological and biochemical parameters across the two sub-population samples of HbSS and HbSC individuals. As expected, statistically significant differences were observed for all hematological parameters except reticulocyte count. Biochemical parameters including creatinine, indirect bilirubin, lactate dehydrogenase (LDH) cholesterol and LDH activity were also different across HbSS and HbSC.
 Click to view | Table 1. Hematologic and Biochemical Parameters of Patients With Hemoglobin SS and SC From Manaus, Amazon, Brazil |
The CAR/CAR haplotype was the most frequent in HbSS individuals (73, 52.5%), followed by CAR/Benin (33, 23.7%), Benin/Benin (25, 18%), CAR/Senegal (4, 2.9%), Benin/Senegal (2, 1.4%), and CAR/Cameroon (2, 1.4%). Among the HbSC individuals, the haplotype distribution was CAR/CI (4, 36.3%), Benin/CI (3, 27.3%), CAR/CII (2, 18.2%), CAR/CIII (1, 9.1%), and Benin/CII (1, 9.1%). Due to the small number of HbSC individuals, the distribution of hematological and biochemical parameters was analyzed only for the HbSS sub-sample (Table 2). When comparing the hematological data among the most frequent haplotypes found in our study, only the MCV presented statistical significance, and the CAR/Ben haplotype presented higher concentration (Fig. 1).
Click to view | Table 2. Hematologic Parameters Between Haplotypes Linked to Globin βS Gene of Patients With Sickle Cell Disease From Manaus, Amazon, Brazil |
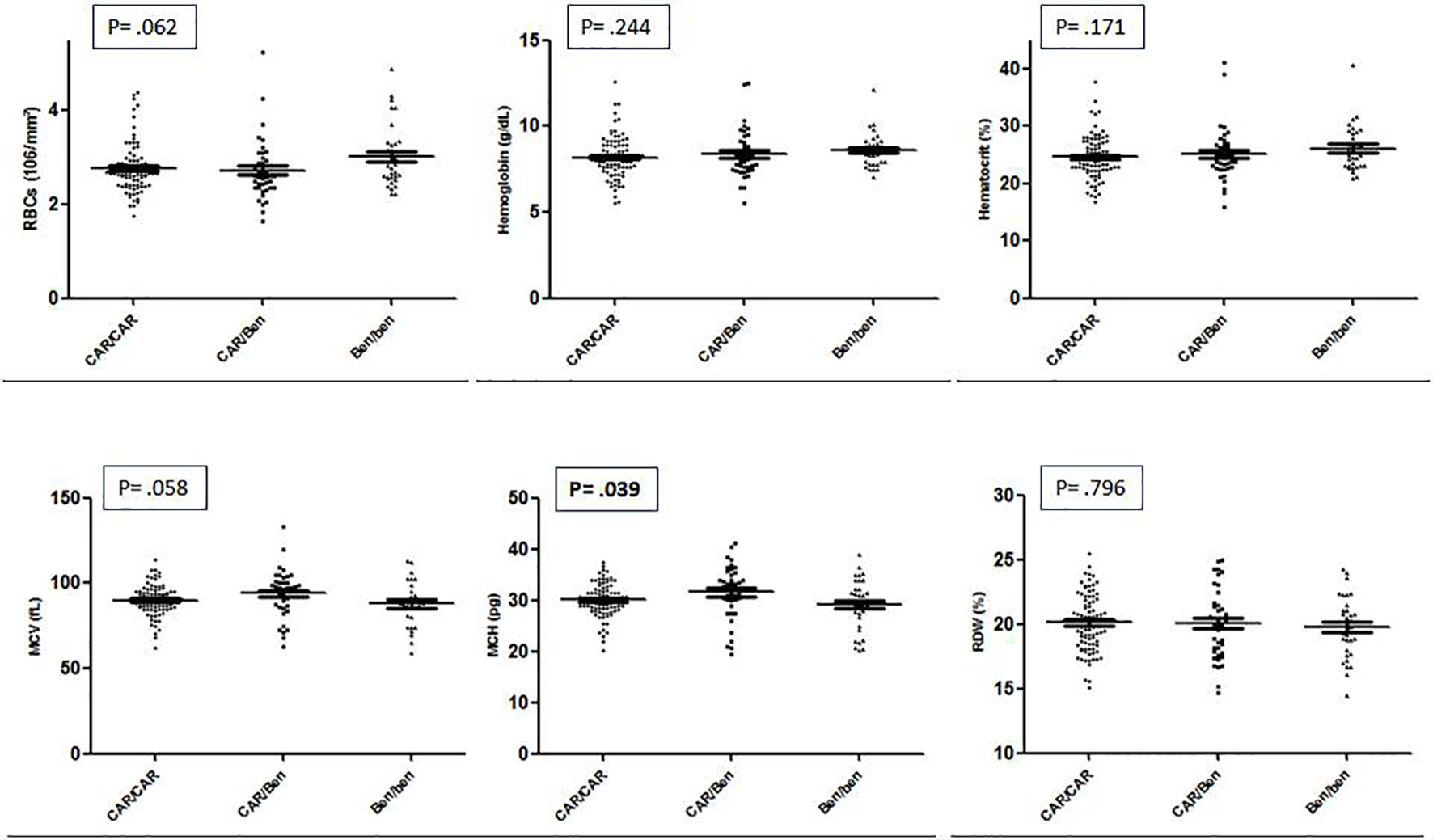 Click for large image | Figure 1. Hematologic parameters between haplotypes CAR/CAR, CAR/Ben and Ben/Ben linked to globin βS gene of patients with sickle cell disease from Manaus, Amazon, Brazil. |
A clear, statistically significant difference was observed for the HbF distribution, with heterozygous CAR/Ben and CAR/Sen presenting average levels of HbF twice and four times as higher that CAR/CAR, respectively. In addition, it is interesting to note that the leucocyte count is reduced among the four individuals CAR/Senegal. Finally, borderline significant differences were detected for RBC count, hematocrit and platelet count.
The single biochemical parameter that showed differential distribution across the haplotype groups was direct bilirubin that was significantly lower (P = 0.029) in the CAR/Cameroon group (0.45 ± 0.07) as compared to CAR/CAR (0.88 ± 0.35), CAR/Ben (0.82 ± 0.38), CAR/Sen (0.70 ± 0.21), Ben/Ben (1.17 ± 0.68) and Ben/Sen (0.78 ± 0.59).
The α-thalassemia 3.7 kb deletion was detected at a frequency of 16.5% in the HbSS group. Of these, 13.7% (19) were heterozygote’s (-α/α) and 2.8% (4) were homozygote’s (-α/-α). Table 3 summarizes the distribution of hematological parameters across two groups of HbSS patients presenting (carriers, both homo- and heterozygous) or not (wild type) the α-thalassemia 3.7 kb deletion. None of the patients SC presented the α-thalassemia 3.7 kb deletion. Statistically significant differences between the two groups were observed for RBC count, hemoglobin, hematocrit, MCV and MCH.
Click to view | Table 3. Analysis of Hematologic Data in Patients of Profile SCD Carrier and Wide Type α-Thalassemia 3.7 kb Deletion From Manaus, Amazon, Brazil |
| Discussion | ▴Top |
The hematologic correlations between HbSS and HbSC patients included in this survey (Table 1) showed a classic profile of laboratory findings associated to sickle cell anemia and SCD. In HbSS patients, there is severe normocytic normochromic anemia associated with reticulocytosis and leukocytosis; HbSC patients presented much milder hematological changes, if any. Of note, LDH activity was above the reference interval on both groups and significantly higher in HbSS patients, suggesting intense hemolysis. Parameters such as MCV, MCH and RDW were higher in HbSS patients, corroborating surveys such as performed by Colella et al in patients with SCD attended in Hematologic and Center of Hematology and Hemotherapy (Sao Paulo, Brazil). In addition, a more pronounced leukocytosis among HbSS patients confirms the participation of leukocytes in the pathophysiology of the disease, likely due to the participation in vaso-occlusive events and not necessarily in infectious processes [27].
Several haplotypes distribution analyses of variants linked to the globin βS gene have been published using population samples from distinct Brazilian regions, with conflicting results. Most of these studies show a predominance of CAR as compared to Ben (the two most frequent genotypes the same result observed here [16, 20, 23, 28]. However, studies performed in population samples from northern Brazilian cities of Salvador and Fortaleza resulted in a predominance of Ben over CAR [12, 19, 21, 29, 30]. Two previous surveys have been published using population samples of βS individuals resident in the Brazilian Amazonic region. The first was performed using a population sample from Belem, capital city of Para, with results somewhat distinct from the present study: three major African haplotypes (CAR, Benin and Senegal) have been identified among 30 SCD patients, with frequencies as follows: CAR/CAR 43%, CAR/Benin 47%, Ben/Ben 7% and Sen/Sen 3%. Sixty-seven percent of the βS chromosomes analyzed were of the CAR type, 30% of the Benin and 3% Senegal [17]. The second focused on African descendants with SCD from three small communities from the Brazilian northern states of Para and Amapa (Curiau, Pacoval and Trombetas) and revealed an even more pronounced predominance of the CAR genotype (60%) as compared to Sen (30%) and Ben (10%) [17, 18]. These discrepancies observed across Brazilian studies involving population samples from different - and even the same - geographic region may be explained by differences in study design (mainly sample sizes) and patterns of migration of Africans to Brazil during the slavery period, and reinforce the need for local surveys.
Results of the comparative analysis of hematological and biochemical parameters across haplotype groups reveal an expected protection of the Benin and Senegal genotypes as compared to CAR, mainly due to a correlation with the levels of HbF, significantly higher in Ben/Sen haplotypes (Table 2) [9, 31, 32]. Interestingly, heterozygous CAR/Ben and CAR/Sen present average levels of HbF twice and four times as higher that CAR/CAR, respectively, suggesting a direct relationship between these variables. Importantly, this must be interpreted with caution due to the small number of CAR/Sen individuals in our sample.
Coexistence of the α-thalassemia 3.7 kb deletion with SCD promotes increase of the erythrocyte’s membrane/cytoplasm ratio with reduced electrolyte loss and cellular dehydration, reduced hemolysis, increase in the concentration of hemoglobin and hematocrit, decreased erythrocyte indices (MCV and MCH) and the reticulocyte count, as shown in our sample [33, 34]. The frequency of the 3.7 kb deletion of our sample of HbSS individuals does not differ from previous Brazilian studies [30, 35]. Our results confirm Rumaney et al that observed an increase in the amounts of RBCs and hemoglobin and reduced MCV in SCD patients also harboring the α-thalassemia 3.7 kb deletion [36]. Pandey et al showed an increase in all parameters (erythrocytes, hemoglobin, hematocrit, MCV and MCH) in HbSS patients with α-thalassemia [37].
In summary, our results show the distribution of βS and βC haplotypes and of α-thalassemia 3.7 kb deletion in a large population sample of SCD patients from the Occidental Brazilian Amazon and the impact of these genotypes over hematological and biochemical parameters, contributing to the expansion of knowledge about the molecular characterization of these diseases in Brazilian populations.
Acknowledgments
The authors wish to thank all patients with sickle cell disease attending the FHEMOAM for contributing and participating in this study. Also, the authors thank the staff of the Molecular Biology Laboratory of the Federal University of Amazonas (UFAM) and from the State University of Amazonas (UEA) for the technical support. Finally, the authors thank the Foundation for Research Support of the State of Amazonas (FAPEAM) for financial support. The sponsors of this study are public or non-profit organizations that support science in general. They had no role in gathering, analyzing or interpreting the data.
Conflicts of Interest
The authors report no conflicts of interest.
Sponsorships
Fundacao de Amparo a Pesquisa do Estado do Amazonas (FAPEAM) - Processo: 1094/2013-FAPEAM.
| References | ▴Top |
- Ingram VM. Abnormal human haemoglobins. III. The chemical difference between normal and sickle cell haemoglobins. Biochim Biophys Acta. 1959;36:402-411.
doi - Bunn HF, Noguchi CT, Hofrichter J, Schechter GP, Schechter AN, Eaton WA. Molecular and cellular pathogenesis of hemoglobin SC disease. Proc Natl Acad Sci U S A. 1982;79(23):7527-7531.
doi pubmed - Weatherall DJ, Clegg JB. Inherited haemoglobin disorders: an increasing global health problem. Bull World Health Organ. 2001;79(8):704-712.
pubmed - Miller AC, Gladwin MT. Pulmonary complications of sickle cell disease. Am J Respir Crit Care Med. 2012;185(11):1154-1165.
doi pubmed - Di Nuzzo DV, Fonseca SF. [Sickle cell disease and infection]. J Pediatr (Rio J). 2004;80(5):347-354.
doi - Nagel RL, Fabry ME, Steinberg MH. The paradox of hemoglobin SC disease. Blood Rev. 2003;17(3):167-178.
doi - Angulo IL, Picado SBR. Hemoglobina C em homozigose e interacao com talassemia beta. Rev Bras Hematol Hemoter. 2009;31(6):408-412.
doi - Pagnier J, Mears JG, Dunda-Belkhodja O, Schaefer-Rego KE, Beldjord C, Nagel RL, Labie D. Evidence for the multicentric origin of the sickle cell hemoglobin gene in Africa. Proc Natl Acad Sci U S A. 1984;81(6):1771-1773.
doi pubmed - Steinberg MH, Nagel RL, Lawrence C, Swaminathan V, Lu ZH, Plonczynski M, Harrell A. Beta-globin gene haplotype in Hb SC disease. Am J Hematol. 1996;52(3):189-191.
doi - Tarer V, Etienne-Julan M, Diara JP, Belloy MS, Mukizi-Mukaza M, Elion J, Romana M. Sickle cell anemia in Guadeloupean children: pattern and prevalence of acute clinical events. Eur J Haematol. 2006;76(3):193-199.
doi pubmed - Nadkarni A, Phanasgaonkar S, Colah R, Mohanty D, Ghosh K. Prevalence and molecular characterization of alpha-thalassemia syndromes among Indians. Genet Test. 2008;12(2):177-180.
doi pubmed - Goncalves MS, Bomfim GC, Maciel E, Cerqueira I, Lyra I, Zanette A, Bomfim G, et al. BetaS-haplotypes in sickle cell anemia patients from Salvador, Bahia, Northeastern Brazil. Braz J Med Biol Res. 2003;36(10):1283-1288.
doi pubmed - Fabry ME, Mears JG, Patel P, Schaefer-Rego K, Carmichael LD, Martinez G, Nagel RL. Dense cells in sickle cell anemia: the effects of gene interaction. Blood. 1984;64(5):1042-1046.
pubmed - Cabral CH, Serafim ES, de Medeiros WR, de Medeiros Fernandes TA, Kimura EM, Costa FF, de Fatima Sonati M, et al. Determination of beta haplotypes in patients with sickle-cell anemia in the state of Rio Grande do Norte, Brazil. Genet Mol Biol. 2011;34(3):421-424.
doi pubmed - Zago MA, Figueiredo MS, Ogo SH. Bantu beta s cluster haplotype predominates among Brazilian blacks. Am J Phys Anthropol. 1992;88(3):295-298.
doi pubmed - Goncalves MS, Nechtman JF, Figueiredo MS, Kerbauy J, Arruda VR, Sonati MF, Saad SO, et al. Sickle cell disease in a Brazilian population from Sao Paulo: a study of the beta s haplotypes. Hum Hered. 1994;44(6):322-327.
doi pubmed - Pante-de-Sousa G, Mousinho-Ribeiro R de C, Guerreiro JF. Origin of the hemoglobin S gene in a northern Brazilian population: the combined effects of slave trade and internal migrations. Genet Mol Biol. 1998;21(4):427-430.
doi - Pante-De-Sousa G, Mousinho-Ribeiro RC, Dos Santos EJ, Guerreiro JF. Beta-globin haplotypes analysis in Afro-Brazilians from the Amazon region: evidence for a significant gene flow from Atlantic West Africa. Ann Hum Biol. 1999;26(4):365-373.
doi pubmed - Galiza Neto GC, Pitombeira MS, Vieira HF, Vieira MLC, Farias DAB. Analysis of βS globin gene haplotypes in Ceara, Brazil. J Bras Patol Med Lab. 2005;41:315-321.
doi - Bezerra MAC, Santos MNN, Araujo AS, Gomes YM, Abath FGC, Bandeira MGC. Molecular variations linked to the grouping of β and α-globin genes in neonatal patients with sickle cell disease in the state of Pernambuco, Brazil. Hemoglobin. 2007;31:1-6.
doi pubmed - Adorno EV, Zanette A, Lyra I, Seixas MO, Reis MG, Goncalves MS. Clinical and molecular characteristics of sickle cell anemia in northeast of Brazil. Genet Mol Biol. 2008;31:621-625.
doi - Camilo-Araujo RF, Amancio OM, Figueiredo MS, Cabanas-Pedro AC, Braga JA. Molecular analysis and association with clinical and laboratory manifestations in children with sickle cell anemia. Rev Bras Hematol Hemoter. 2014;36(5):334-339.
doi pubmed - Okumura JV, Lobo CL, Bonini-Domingos CR. Beta-S globin haplotypes in patients with sickle cell anemia: one approach to understand the diversity in Brazil. Rev Bras Hematol Hemoter. 2013;35(1):71-72.
doi pubmed - Betke K, Marti HR, Schlicht I. Estimation of small percentages of foetal haemoglobin. Nature. 1959;184(Suppl 24):1877-1878.
doi pubmed - Sutton M, Bouhassira EE, Nagel RL. Polymerase chain reaction amplification applied to the determination of beta-like globin gene cluster haplotypes. Am J Hematol. 1989;32(1):66-69.
doi pubmed - Baysal E, Huisman TH. Detection of common deletional alpha-thalassemia-2 determinants by PCR. Am J Hematol. 1994;46(3):208-213.
doi pubmed - Colella MP, de Paula EV, Machado-Neto JA, Conran N, Annichino-Bizzacchi JM, Costa FF, Olalla Saad ST, et al. Elevated hypercoagulability markers in hemoglobin SC disease. Haematologica. 2015;100(4):466-471.
doi pubmed - Costa FF, Arruda VR, Goncalves MG, Miranda SR, Carvalho MH, Sonati MF, Saad SO, et al. Beta S-gene-cluster haplotypes in sickle cell anemia patients from two regions of Brazil. Am J Hematol. 1994;45(1):96-97.
doi pubmed - Silva LB, Goncalves RP, Rabenhorst SHB. Analysis of sickle cell anemia haplotypes in Fortaleza reveals the ethnic origins of Ceara state population. J Bras Patol Med Lab. 2009;45:115-118.
doi - Adorno EV, Couto FD, Moura Neto JP, Menezes JF, Rego M, Reis MG, Goncalves MS. Hemoglobinopathies in newborns from Salvador, Bahia, Northeast Brazil. Cad Saude Publica. 2005;21(1):292-298.
doi pubmed - Powars DR. Beta s-gene-cluster haplotypes in sickle cell anemia. Clinical and hematologic features. Hematol Oncol Clin North Am. 1991;5(3):475-493.
pubmed - Lu ZH, Steinberg MH. Fetal hemoglobin in sickle cell anemia: relation to regulatory sequences cis to the beta-globin gene. Multicenter Study of Hydroxyurea. Blood. 1996;87(4):1604-1611.
pubmed - Nagel RL, Steinberg MH. Genetics of the βS gene: Origins, epidemiology, and epistasis. In: Steinberg MH, Forget BG, Higgs DR, Nagel X RL, editors. Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management, 1st ed. Cambridge: Cambridge University Press. 2001; p. 711-755.
- Steinberg MH. Predicting clinical severity in sickle cell anaemia. Br J Haematol. 2005;129(4):465-481.
doi pubmed - Cardoso GL, Takanashi SY, Guerreiro JF. Inherited hemoglobin disorders in an Afro-Amazonian community: Saracura. Genet Mol Biol. 2012;35(3):553-556.
doi pubmed - Rumaney MB, Ngo Bitoungui VJ, Vorster AA, Ramesar R, Kengne AP, Ngogang J. The co-inheritance of alpha-thalassemia and sickle cell anemia is associated with better hematological indices and lower consultations rate in Cameroonian patients and could improve their survival. PLoS One. 2014;9:1-10.
doi pubmed - Pandey S, Mishra RM, Sharma M, Saxena R. Genotypic influence of alpha-deletions on the phenotype of Indian sickle cell anemia patients. Korean J Hematol. 2011;46(3):192-195.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.