

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website http://www.thejh.org |
Original Article
Volume 7, Number 2, May 2018, pages 62-68
Protein C and Anti-Thrombin-III Deficiency in Children With Beta-Thalassemia
Suzy Abd El Mabooda, e, Doaa Moawad Fahmyb, Ahmed Akefc, Shadia El Sallabd
aHematology, Oncology and Bone Marrow Transplantation Unit, Pediatric Department, Faculty of Medicine, Mansoura University, Egypt
bPediatric Department, Faculty of Medicine, Mansoura University, Egypt
cClinical Pathology Department, Faculty of Medicine, Mansoura University, Egypt
dNeonatal Intensive Care Unit, Pediatric Department, Faculty of Medicine, Mansoura University, Egypt
eCorresponding Author: Suzy Abd El Mabood, Hematology, Oncology and Bone Marrow Transplantation Unit, Pediatric Department, Faculty of Medicine, Mansoura University, Egypt
Manuscript submitted March 26, 2018, accepted April 30, 2018
Short title: Protein C and Anti-Thrombin-III in Thalassemia
doi: https://doi.org/10.14740/jh392w
| Abstract | ▴Top |
Background: Thromboembolic events (TEEs) are recently described complications in thalassemia patients. Many mechanisms were postulated for thrombosis. Conflicting results of natural anticoagulants values were reported in previous studies. Our aim was to investigate protein C and anti-thrombin-III (AT-III) levels in thalassemics and to detect risk factors for their decrement.
Methods: A cross-sectional study for 60 beta-thalassemia patients (35 major and 25 intermedia) and 35 healthy children were tested for protein C and AT-III levels, liver function tests and Sr. ferritin.
Results: A significant reduction in protein C and AT-III levels was noticed in patient group compared to healthy children (82.50% (32 - 175) vs. 104% (60 - 204), P = 0.041 and 237.52 ± 53.19 mg/L vs. 322.99 ± 56.57 mg/L, P value ≤ 0.001, respectively). Protein C was lower among older patients (> 10 years) than younger patients (< 10 years), and splenectomized category than non-splenectomized one (P = 0.02 and 0.011, respectively). AT-III was significantly lower among splenectomized patients as compared to those who did not undergo splenectomy (P = 0.04). Significant correlations were found between protein C and AT-III with older age and liver functions.
Conclusions: Protein C and AT-III were significantly lower among thalassemics with the main risk factors for their deficiencies being: splenectomy and increasing age. This allows establishment of early prophylactic policy against TEE for the vulnerable group.
Keywords: Anti-thrombin-III; Protein C; Thalassemics; Thromboembolic events
| Introduction | ▴Top |
A dramatic improvement has been noticed in the management of thalassemia patients over the last 10 - 20 years with resulting increase in their life expectancy [1]. This is thanks to advances in methods of both evaluation and treatment of iron overload with the availability of different iron chelation therapies [1], yet more complications have emerged [2].
Thromboembolic events (TEEs) secondary to hypercoagulable state are relatively more recent complications being identified among beta-thalassemia patients [3].
Many mechanisms were postulated to be behind TEE in thalassemics such as: splenectomy, low hemoglobin levels, increasing age, presence of comorbidities as cardiac, hepatic, or endocrinal dysfunctions [4], transfusion naivety [3], low levels of anticoagulants (e.g. protein C and protein S), increased production of thrombin, platelet activation and enhanced aggregation stimulated by thalassemic RBCs [5, 6].
Values of naturally occurring anticoagulants in beta-thalassemia patients are suggested to be low with increased risk of TEE [5-8]. Nevertheless, such issue is still debatable among researchers [9, 10]. While some found low levels of natural anticoagulants [7, 8], others found normal values [9, 10]. In this study, we investigated the level of anti-thrombin-III (AT-III) and protein C among a group of beta-thalassemia children and correlated to different clinical and laboratory features in order to help hematologists to establish of a prophylactic policy for each patient according to his risk.
| Materials and Methods | ▴Top |
Study population
This study was a case-control study carried out in Mansoura Universtiy Children’s Hospital, Mansoura, Egypt. Sixty beta-thalassemia patients including 35 thalassemia major (TM) and 25 thalassemia intermedia (TI) (28 males and 32 females) with an age range from 2.5 to 17 years from Pediatric Hematology Outpatient Clinic were enrolled in this study. The diagnosis of beta-thalassemia was based upon the results of hemoglobin electrophoresis and the clinical course of the disease. TI was distinguished from TM when the diagnosis was made after the age of 2 years old and transfusion frequency was less than 12 per year, or if the patients maintained hemoglobin of 7 g/dL without transfusion. Two groups of patients were included in this study: splenectomized and non-splenectomized groups and they were selected sequentially in the order of 1:1. Also 35 age- and sex-matched healthy children were involved for comparison. The study did not include patients with other forms of chronic hemolytic anemias (e.g. sickle thalassemia) or those suffering from severe hepatic or cardiac dysfunctions. Also patients with history of familial thrombophilia or use of anticoagulant therapy were excluded.
Informed consents/ascents were obtained from patients and/or their guardians before initiating the study in accordance with Declaration of Helsinki. The study protocol was approved before conducting the study by Institutional Review Board of Faculty of Medicine, Mansoura University.
The patients were evaluated clinically for: duration of the disease, age at diagnosis, frequency of blood transfusion, date of the last transfusion, anti-platelet drug intake, history of splenectomy, any symptoms suggestive of TEE, and family history of TEE. Also patients were examined for features of decompensated hepatic or cardiac diseases or signs suggestive for thrombosis.
Laboratory investigations were done for the following tests: CBC, hemoglobin electrophoresis, liver function tests, serum creatinine, virology screening, serum ferritin, prothrombin time and international normalized ratio (INR).
Protein C measurement
It was measured by REAADS® protein C antigen test kit read by Stat Fax. It was expressed as relative % in comparison to pooled normal plasma. The normal values were 72-160%.
AT-III measurement
Spinreact kits read by Stat Fax were used for its measurement. The normal values were between 200 and 400 mg/L.
Statistical analysis
Data were analyzed using SPSS version 21 (Statistical Package for the Social Sciences, SPSS Inc., Chicago, IL, USA). Continuous variables were presented as mean ± standard deviation (SD) for quantitative parametric data and median (minimum and maximum) for quantitative non-parametric data. Comparison was made between two independent mean groups for parametric data using Student’s t-test. Comparison was made between two independent groups for non-parametric data using Mann-Whitney test. Ranked Spearman correlation test was used to study the possible association between variables for non-parametric data. Pearson correlation was used for correlation between continuous parametric data. Chi-square test was used to study the association between each two variables or comparison between two independent groups as regards the categorized data. Results were considered significant when the probability of error is less than 5% (P < 0.05).
| Results | ▴Top |
Clinical and laboratory characteristics of the patient group were shown in Table 1. Splenomegaly were found in 23 (76%) of non-splenectomized patients; 31.6% (19/60) thalassemia patients were HCV infected, and 12/30 (40%) patients were maintained on low dose aspirin as post splenectomy antiplatelet medication. None of the patients experienced clinically manifested thrombosis.
 Click to view | Table 1. Clinical and Laboratory Characteristics of Thalassemia Patients Group |
A significant reduction in protein C and AT-III levels was noticed in patient group when compared to those of healthy children (Figures 1 and 2) (82.50% (32 - 175) vs. 104% (60 - 204), P = 0.041 and 237.52 ± 53.19 mg/L vs. 322.99 ± 56.57 mg/L, P ≤ 0.001, respectively). Protein C deficiency was observed in 13/60 patients (21.7%) vs. 2/35 controls (Fig. 3). AT-III was low in 12/60 patients (20%), while none of the controls had low AT-III value (Fig. 4) (Table 2).
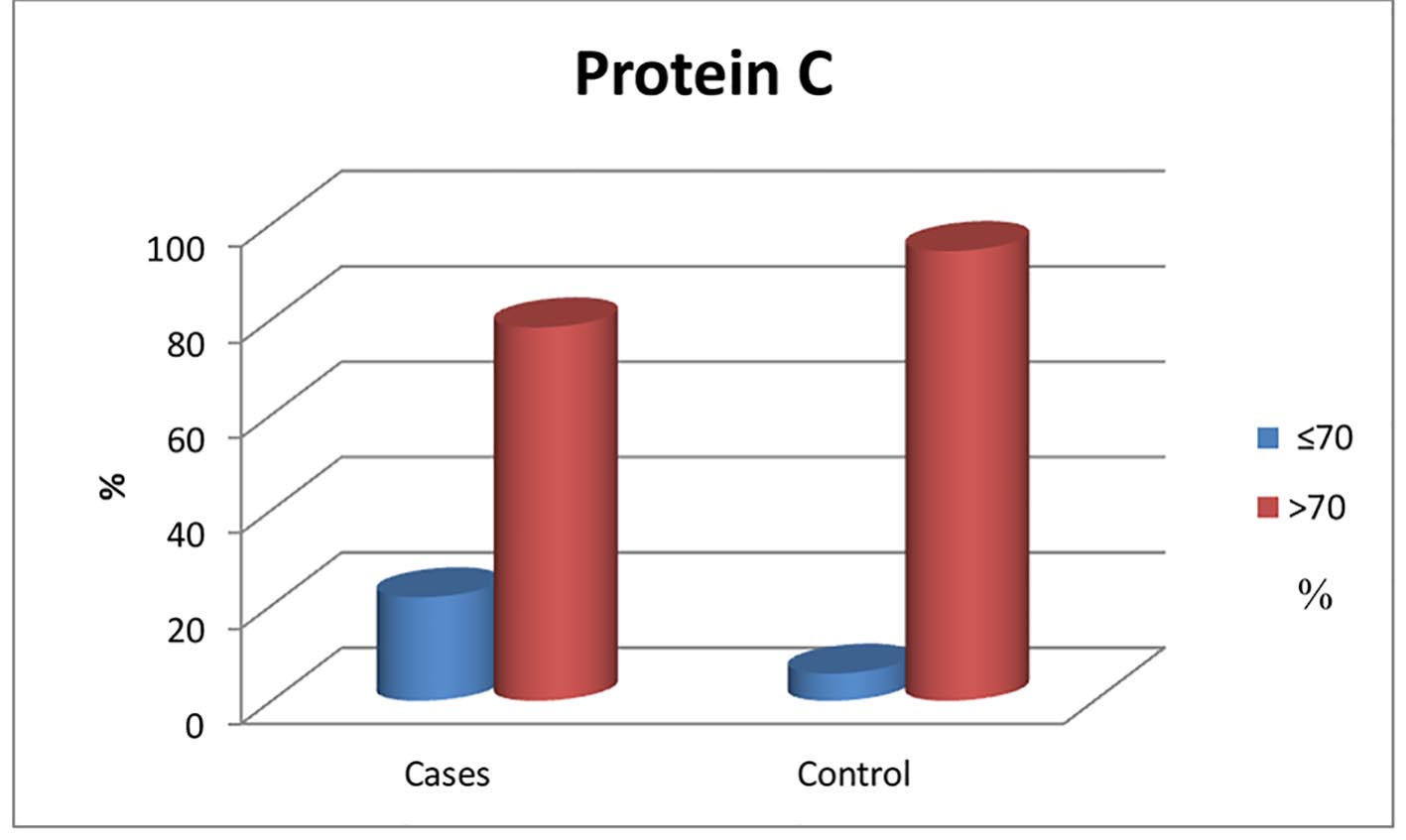 Click for large image | Figure 1. Normal and below normal protein C levels in patients and control group. |
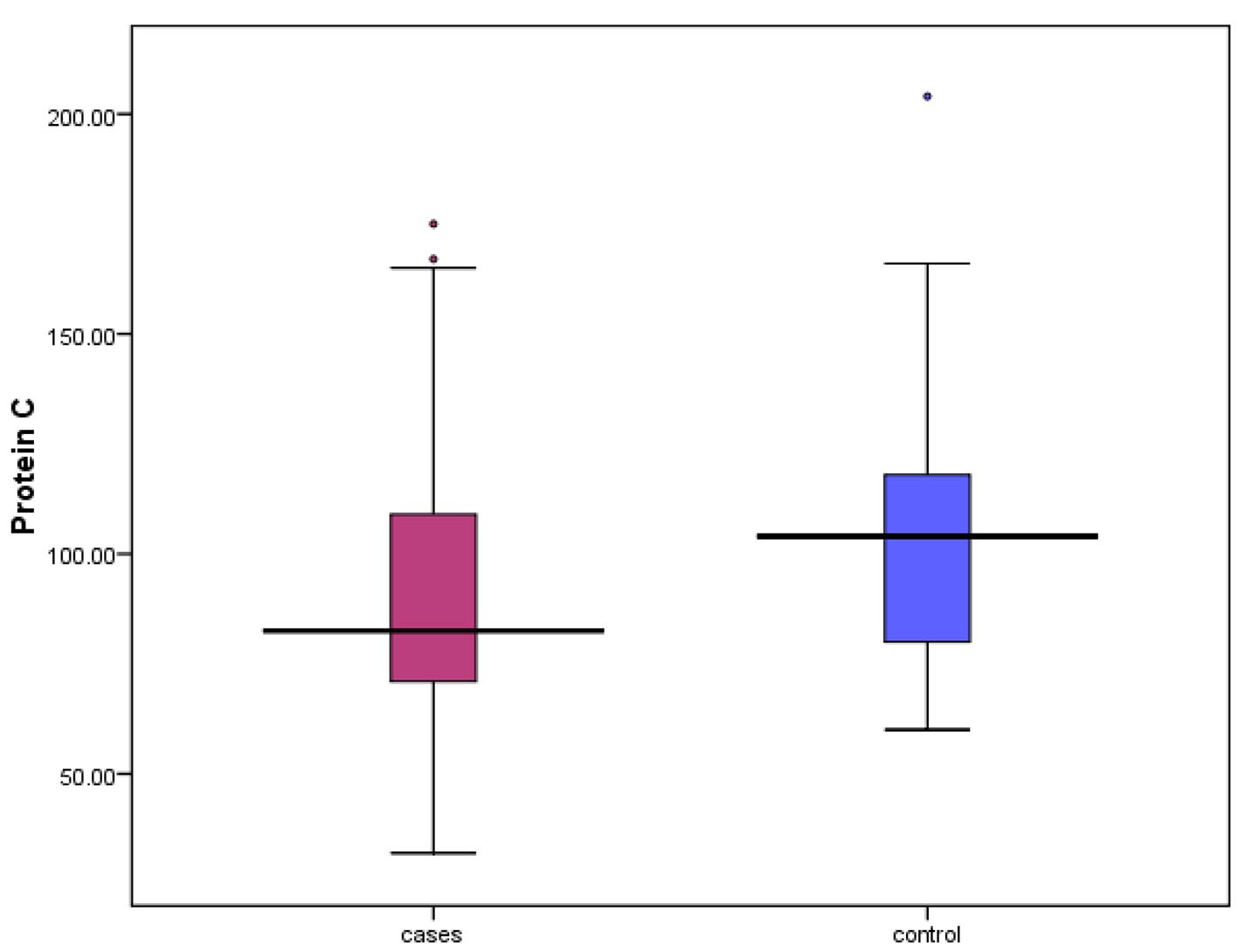 Click for large image | Figure 2. Median levels of protein C in both patients and control. |
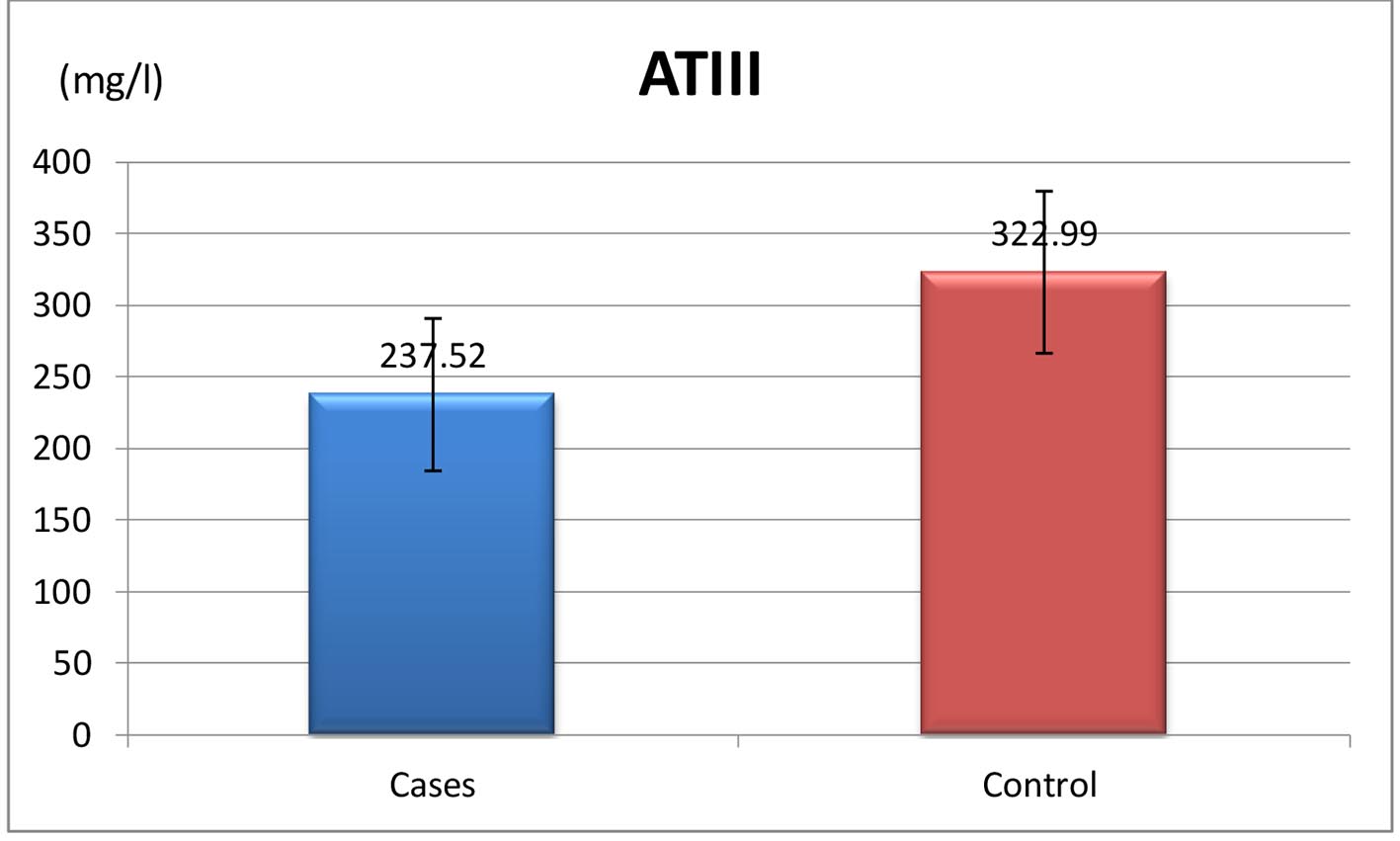 Click for large image | Figure 3. Mean values of AT-III in both patients and control group. |
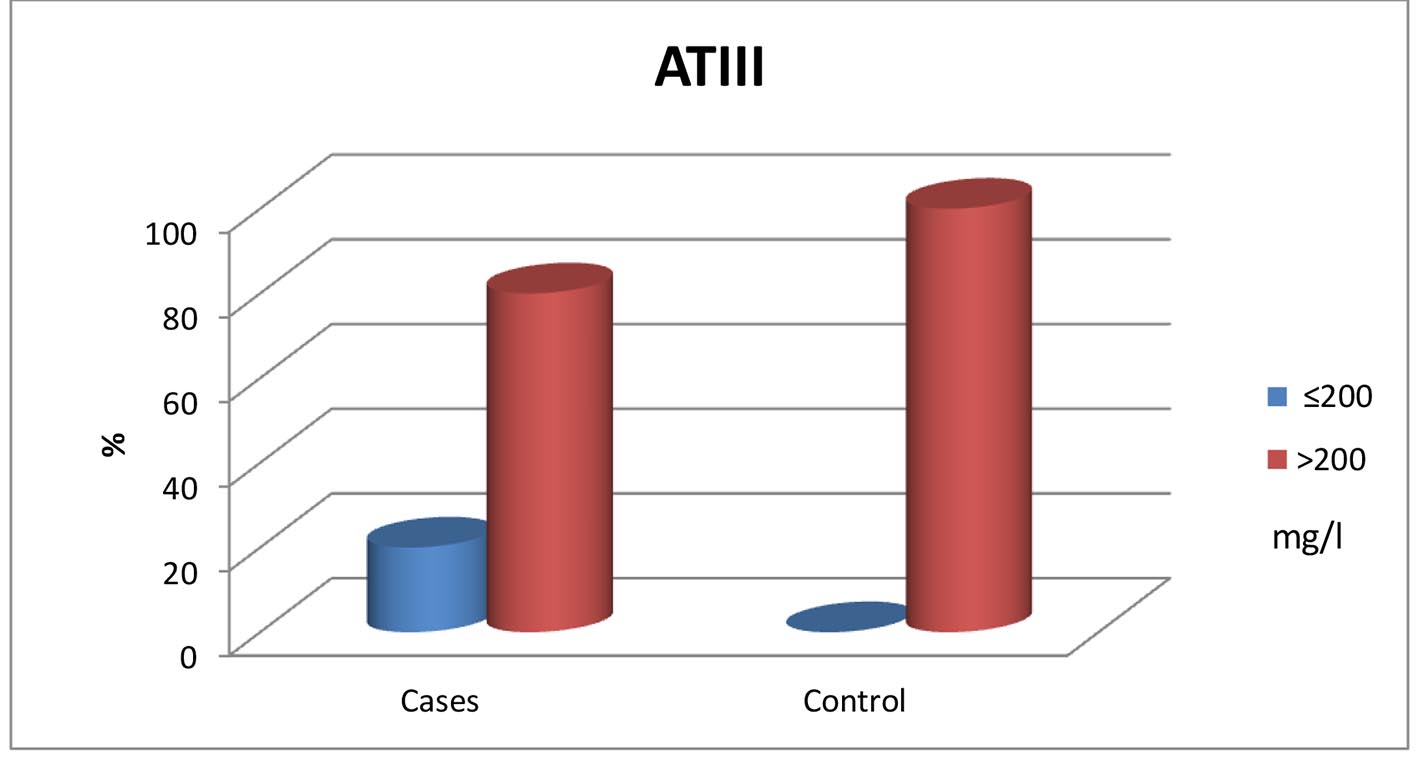 Click for large image | Figure 4. Normal and below normal values of both patients and control groups. |
Click to view | Table 2. Protein C and AT-III in Both Patients and Healthy Groups |
Levels of protein C and AT-III were compared to different patients’ clinical and laboratory criteria (Table 3). A significant reduction was found in protein C level among older patients (> 10 years old) and splenectomized category (P = 0.02 and 0.011, respectively). For AT-III, significant differences were noticed among splenectomized patients (P = 0.04).
Click to view | Table 3. Protein C and AT-III in Comparison to Other Clinical Laboratory Criteria |
Positive correlations between both protein C and AT-III with Sr. albumin were found. However, statistically significant negative correlations between protein C and patients’ age, ALT, Sr. direct bilirubin, and INR were detected. AT-III was negatively correlated with patients’ age, ALT, AST, Sr. direct bilirubin, and INR (Table 4).
Click to view | Table 4. Correlation Between Protein C, AT-III and Different Clinical and Laboratory Characteristics of the Patients Group |
| Discussion | ▴Top |
Hypercoagulable state and consequently TEE are relatively more recent complications among beta-thalassemia patients which may result in significant morbidity and mortality [3]. Such problem is not only limited to TI patients but activation of several hemostatic mechanisms have been also described in TM individuals [11]. Different risk factors were suggested to be beyond TEE in thalassemia albeit splenectomy and transfusion naivety are the most highlighted leading causes [12, 13].
In this study, we compared natural anticoagulants levels (protein C and AT-III) in β-thalassemia patients to their healthy peers. In our study, only 13% and 12% of the patients reported below normal protein C and AT-III levels, respectively compared to healthy children. Previous studies reported similar results [11, 14-16], but with variable percentages of affected individuals as Abosdera et al [14] reported 56% of thalassemia patients had low protein C and 54% with lower AT-III than normal.
Many mechanisms were speculated to be behind low levels of naturally occurring anticoagulants in thalassemia patients. As protein C and AT-III are synthesized in the liver, they are very sensitive to any synthetic function defect which is common among those patients because of many offending factors such as: hemosiderosis and viral hepatitis [10, 11, 17-19]. This was supported by the significant correlations between levels of anticoagulants and synthetic function of the liver (albumin and INR values). Researchers think that liver impairment is not the only explanation behind their deficiency [20]. Another reason for this is their entrapment on RBCs membrane by negatively charged phospholipids particularly phosphatidylserine which are abnormally found in the external membrane of the thalassemic RBCs [21]. Furthermore, some authors assumed increased anticoagulant consumption in thalassemia as a result of chronic subclinical activation of coagulation system evidenced by higher thrombin-antithrombin (TAT) complexes level detected among TM patients compared to normal [10, 22].
Protein C and AT-III were compared to various patients’ features in an attempt to identify who are at greater risk for developing hypercoagulable state. Both protein C and AT-III were significantly lower among splenectomized category when compared to non-splenectomized one.
The explanation for that may be the presence of procoagulants on the surface of RBCs and abnormal platelets which are no longer removed from the circulation after splenectomy with the resultant increase in protein C consumption to control the hypercoagulable state [23, 24]. Previous publications did not reach a uniform agreement regarding this point. While some researchers found similar results to ours [22, 25], others found comparable levels when compared to normal individuals [8].
The reduction in protein C level was more pronounced among older patients. Musumeci et al [26] found similar finding as the lowest protein C values were observed among older splenectomized patients. Although we did not find the same result with AT-III, it was negatively correlated with age. Taher et al [27] examined the data of 8,860 thalassemia patients from the Mediterranean area and Iran and found the old age (above 20 years) was one of the major risk factors for the development of TEE. This also may explain why none of our patients has experienced clinically overt TEE as the age of the study group is still younger than the risky age for TEE development. Moreover, Taher et al [28] studied how long it took for a thrombosis to develop after splenectomy and the median time to thrombosis was 8 years suggesting that TEE is not an acute complication for splenectomy; it is more likely to be a manifestation of a chronic underlying process.
No significant differences could be noticed between males and females regarding anticoagulants levels denoting that both sexes are at equal risk for TEE development. Apart from Karami et al [25] reporting lower levels among males, other studies did not find significant sex differences [27]. Although TEEs were reported much more in TI than TM patients [2, 27], we could not find a significant difference in anticoagulants levels between both groups. Perhaps this is owing to the presence of many contributing factors for the development of TEE in TI other than deficiency of natural anticoagulant levels. Furthermore, sometimes there is an overlap in clinical presentations between both TM and TI making the distinction from each other difficult. Although prophylactic anticoagulants are indicated for TI patients exposed to thrombotic risk factors [28], they are recommended only for TM patients when thromboembolic complications occur. Further studies are warranted to determine whether lifelong use of prophylactic anticoagulants is needed to prevent subclinical thrombosis in the lungs and brain or not [6]. More clinical laboratory data about patients who are at risk of hypercoagulability help in establishing a prophylactic plan against thrombosis for each patient according to his risk.
Conclusion
Naturally occurring anticoagulants (protein C and AT-III) were significantly lower among thalassemia children especially among splenectomized and older aged patients. This may suggest the need for further evaluation or even early prophylactic policy against TEE in these patients.
Acknowledgments
We would like to thank our patients, their parents, nurses and physicians in Mansoura University Children Hospital, Mansoura, Egypt for their trust. Without their cooperation this work could not be accomplished.
Conflict of Interest
None of the authors has any conflict of interest.
Author Contributions
Suzy Abd El Mabood was the owner of the idea, established the study design and shared in writing the manuscript. Doaa Moawad Fahmy collected the clinical and laboratory data, analyzed and interpreted the results. Ahmed Akef did the laboratory tests and revised manuscript. Shadia El Sallab supervised all steps of the work and revised the final copy of the manuscript.
Financial Disclosure
No relevant financial interests.
Funding Support
None.
| References | ▴Top |
- Musallam KM, Rivella S, Vichinsky E, Rachmilewitz EA. Non-transfusion-dependent thalassemias. Haematologica. 2013;98(6):833-844.
doi pubmed - Taher AT, Musallam KM, Karimi M, El-Beshlawy A, Belhoul K, Daar S, Saned MS, et al. Overview on practices in thalassemia intermedia management aiming for lowering complication rates across a region of endemicity: the OPTIMAL CARE study. Blood. 2010;115(10):1886-1892.
doi pubmed - Cappellini MD, Motta I, Musallam KM, Taher AT. Redefining thalassemia as a hypercoagulable state. Ann N Y Acad Sci. 2010;1202:231-236.
doi pubmed - Taher AT, Otrock ZK, Uthman I, Cappellini MD. Thalassemia and hypercoagulability. Blood Rev. 2008;22(5):283-292.
doi pubmed - Butthep P, Rummavas S, Wisedpanichkij R, Jindadamrongwech S, Fucharoen S, Bunyaratvej A. Increased circulating activated endothelial cells, vascular endothelial growth factor, and tumor necrosis factor in thalassemia. Am J Hematol. 2002;70(2):100-106.
doi pubmed - Eldor A, Rachmilewitz EA. The hypercoagulable state in thalassemia. Blood. 2002;99(1):36-43.
doi pubmed - Cappellini MD, Musallam KM, Marcon A, Taher AT. Coagulopathy in Beta-thalassemia: current understanding and future perspectives. Mediterr J Hematol Infect Dis. 2009;1(1):e2009029.
doi - Hashemieh M, Azarkeivan A, Sheibani K. A comparison of hemostatic changes in splenectomized and nonsplenectomized beta-thalassemia intermedia patients. J Pediatr Hematol Oncol. 2016;38(8):636-641.
doi pubmed - Rosnah B, Halina N, Shafini Y, Marini R, Rosline H. The level of natural anticoagulants in transfusion dependent thalassemia patients in Kelantan, Northeastern Malaysia. J Hematol Thromb Dis. 2014;2:2-5.
- Chhikara A, Sharma S, Chandra J, Nangia A. Thrombin Activable Fibrinolysis Inhibitor in Beta Thalassemia. Indian J Pediatr. 2017;84(1):25-30.
doi pubmed - Hassan TH, Elbehedy RM, Youssef DM, Amr GE. Protein C levels in beta-thalassemia major patients in the east Nile delta of Egypt. Hematol Oncol Stem Cell Ther. 2010;3(2):60-65.
doi - Succar J, Musallam KM, Taher AT. Thalassemia and venous thromboembolism. Mediterr J Hematol Infect Dis. 2011;3(1):e2011025.
doi pubmed - Cappellini MD, Musallam KM, Poggiali E, Taher AT. Hypercoagulability in non-transfusion-dependent thalassemia. Blood Rev. 2012;26(Suppl 1):S20-23.
doi - Abosdera MM, Almasry AE, Abdel-Moneim ES. Coagulation defects in thalassemic patients. Pediatr Neonatol. 2017;58(5):421-424.
doi pubmed - Sipahi T, Kara A, Kuybulu A, Egin Y, Akar N. Congenital thrombotic risk factors in beta-thalassemia. Clin Appl Thromb Hemost. 2009;15(5):581-584.
doi pubmed - Shebl SS, el-Sharkawy HM, el-Fadaly NH. Haemostatic disorders in nonsplenectomized and splenectomized thalassaemic children. East Mediterr Health J. 1999;5(6):1171-1177.
pubmed - Cappellini MD, Grespi E, Cassinerio E, Bignamini D, Fiorelli G. Coagulation and splenectomy: an overview. Ann N Y Acad Sci. 2005;1054:317-324.
doi pubmed - Naithani R, Chandra J, Narayan S, Sharma S, Singh V. Thalassemia major—on the verge of bleeding or thrombosis? Hematology. 2006;11(1):57-61.
doi pubmed - Mustafa NY, Marouf R, Al-Humood S, Al-Fadhli SM, Mojiminiyi O. Hypercoagulable state and methylenetetrahydrofolate reductase (MTHFR) C677T mutation in patients with beta-thalassemia major in Kuwait. Acta Haematol. 2010;123(1):37-42.
doi pubmed - Shirahata A, Funahara Y, Opartkiattikul N, Fucharoen S, Laosombat V, Yamada K. Protein C and protein S deficiency in thalassemic patients. Southeast Asian J Trop Med Public Health. 1992;23(Suppl 2):65-73.
pubmed - Wilairat P, Kittikalayawong A, Chaicharoen S. The thalassemic red cell membrane. Southeast Asian J Trop Med Public Health. 1992;23(Suppl 2):74-78.
pubmed - Angchaisuksiri P, Atichartakarn V, Aryurachai K, Archararit N, Chuncharunee S, Tiraganjana A, Rattanasiri S. Hemostatic and thrombotic markers in patients with hemoglobin E/beta-thalassemia disease. Am J Hematol. 2007;82(11):1001-1004.
doi pubmed - Elgammal M, Mourad Z, Sadek N, Abassy H, Ibrahim H. Plasma levels of soluble endothelial protein C-receptor in patients with b-thalassemia. Alexandria J Med. 2012;48:283-288.
doi - Ruf A, Pick M, Deutsch V, Patscheke H, Goldfarb A, Rachmilewitz EA, Guillin MC, et al. In-vivo platelet activation correlates with red cell anionic phospholipid exposure in patients with beta-thalassaemia major. Br J Haematol. 1997;98(1):51-56.
doi pubmed - Karami H, Vahidshahi K, Kosarian M, Karami H, Shahmohammadi S, Dabirian M, Vafainezhad M, et al. Assessment of coagulation state and its related factors in thalassemia intermedia patients referred to thalassemia research center at Booali Sina Hospital Sari/IR Iran in 2007. Pak J Biol Sci. 2010;13(9):448-451.
doi pubmed - Musumeci S, Leonardi S, Di Dio R, Fischer A, Di Costa G. Protein C and antithrombin III in polytransfused thalassemic patients. Acta Haematol. 1987;77(1):30-33.
doi pubmed - Taher A, Isma'eel H, Mehio G, Bignamini D, Kattamis A, Rachmilewitz EA, Cappellini MD. Prevalence of thromboembolic events among 8,860 patients with thalassaemia major and intermedia in the Mediterranean area and Iran. Thromb Haemost. 2006;96(4):488-491.
doi pubmed - Cappellini MD, Robbiolo L, Bottasso BM, Coppola R, Fiorelli G, Mannucci AP. Venous thromboembolism and hypercoagulability in splenectomized patients with thalassaemia intermedia. Br J Haematol. 2000;111(2):467-473.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.