

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website http://www.thejh.org |
Case Report
Volume 8, Number 2, June 2019, pages 68-70
Macrophage Activation Syndrome Versus Hemophagocytic Lymphohistiocytosis: A Diagnostic Dilemma in a Patient With Still’s Disease and Epstein-Barr Virus Viremia
Roberta Gomeza, c, Joseph Maakaronb, Robert Baiocchib
aDepartment of Medicine/Pediatrics, The Ohio State University, Columbus, OH 43210, USA
bDivision of Hematology/Oncology, Department of Medicine, The Ohio State University, Columbus, OH 43210, USA
cCorresponding Author: Roberta Gomez, 700 Children’s Drive, ED277, Columbus, OH 43205, USA
Manuscript submitted March 19, 2019, accepted June 6, 2019
Short title: HLH Versus MAS
doi: https://doi.org/10.14740/jh495
| Abstract | ▴Top |
Macrophage activation syndrome (MAS) and hemophagocytic lymphohistiocytosis (HLH) are two overlapping, potentially fatal syndromes classified by disorganization and malfunction of the immune system that results in wide spread inflammation and end-organ damage. We present the case of a 22-year-old female with both underlying adult-onset still’s disease and active Epstein-Barr virus (EVB) viremia who presented with criteria for MAS/HLH. She ultimately improved on an immunosuppressive regimen, and during follow-up was also found to be heterozygote carrier for a known genetic mutation that has been associated with “primary” HLH. This case thus highlights the clinical spectrum of HLH/MAS, the different treatment approaches, and the new investigations into the relationship between primary and secondary HLH.
Keywords: Hemophagocytic lymphohistiocytosis; Macrophage activation syndrome; Adult-onset still’s disease; EBV viremia; Anakinra
| Introduction | ▴Top |
Macrophage activation syndrome (MAS) and hemophagocytic lymphohistiocytosis (HLH) are two overlapping, potentially fatal syndromes classified by disorganization and malfunction of the immune system that results in wide spread inflammation and end-organ damage [1]. MAS is classically associated with rheumatologic conditions such as systemic juvenile idiopathic arthritis (sJIA) and adult-onset Still’s disease (AOSD) [1, 2]. Contrarily, HLH is typically associated with viral infections, malignancy, and certain chemotherapies [1]. Both syndromes can also be seen in patients with mutations of genes involved in the lymphocyte cytolytic pathway [1, 3], in which they are referred to as having primary or familial HLH (FHLH). Although somewhat different in etiology, their manifestations are quite overlapping. Both MAS and HLH present clinically with marked hyperferritinemia, cyclic fevers, cytopenias, hepatosplenomegaly, coagulopathy, and if not treated, end-organ failure and death [3]. The mainstay of therapy for MAS would be control of the underlying rheumatologic disorder with immunosuppressant therapy [2, 4], while the treatment of choice for HLH would likely be a combination of immunosuppressive, anti-viral and/or chemotherapy agents depending upon the trigger. We herein present a case of immune overactivation in a patient with Still’s disease on active immunosuppression.
| Case Report | ▴Top |
A 22-year-old female with a history of migraines and recently diagnosed AOSD presented to the emergency room after initially presenting to an outside hospital with 5 days of daily, high grade fevers and severe lower abdominal pain with diffuse body aches. She endorsed having this type of pain before, at the time of her diagnosis with AOSD 1 year prior. Her immunosuppressive regimen consisted of anakinra 100 mg daily and hydroxychloroquine 400 mg daily.
She was tachycardic and febrile. She had bilateral tonsillar erythema, cervical lymphadenopathy, moderate tenderness to palpation in her right lower quadrant, and a diffuse erythematous papular rash on the dorsum of her shins bilaterally. She had no focal joint tenderness, erythema or edema, but did endorse diffuse tenderness over her trunk and extremities. Lab work revealed elevated transaminases (alkaline phosphate (ALP) 136, aspartate transferase (AST) 241, and alanine transferase (ALT) 161), hyperferritinemia (9,445), elevated lactate dehydrogenase (LDH) 990 and cytopenia (white blood cell (WBC) 3.5, platelets 90,000). Given these lab abnormalities in addition to her daily fevers and lymphadenopathy, she met criteria for MAS.
She was started on high dose intravenous (IV) steroids. EBV PCR revealed 79,162 copies, with a positive IgM titer and negative IgG. Additionally, her soluble CD25 (IL2-receptor) came back elevated at 2,660 (upper limit of 1,033), which then allowed the patient to meet diagnostic criteria for HLH [5]. Thus, it became unclear whether the patient had MAS secondary to an acute flare of AOSD, or whether she had HLH secondary to acute EBV infection, or potentially a combination of both processes. Given the concern for HLH with possible underlying malignancy, she underwent a computed tomography ( CT) of the chest, abdomen and pelvis, which was negative for lymphadenopathy but did confirm splenomegaly. Ultimately, her labs and exam continued to improve (Fig. 1) with high-dose steroids and immunosuppressants, and with the addition of cyclosporine. Her good clinical response to immunosuppressive therapy therefore suggested that her underlying AOSD may have been driving her disease process.
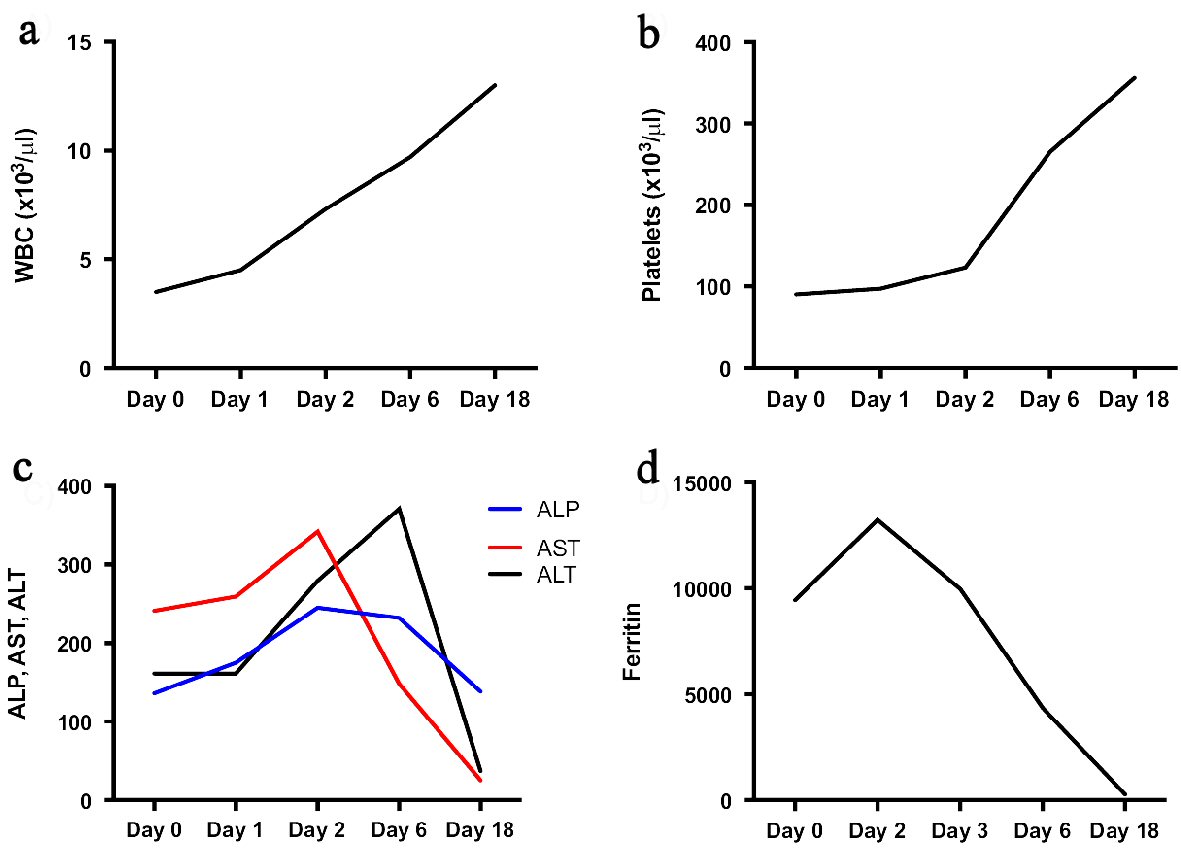 Click for large image | Figure 1. Overall trend in disease-defining labs. This figure demonstrates white blood cell (WBC) (a), platelet (b), alkaline phosphate (ALP), aspartate transferase (AST), and alanine transferase (ALT) (c) and ferritin (d) levels over the course of admission. Of note, Day 0 represents the day of admission, high dose steroids (dexamethasone 40 mg intravenous) were started on Day 1, and cyclosporine 50 mg twice daily was started on Day 5. The patient was discharged on Day 6, and seen in outpatient follow-up on Day 18. |
| Discussion | ▴Top |
There was much consideration about initiating etoposide, as proposed in the HLH 1994/2004 [5-7] criteria for patients who met at least five of the eight criteria for HLH. There was also discussion as to whether rituximab and/or antiviral therapy given the patient’s high EBV viral load [6]. As mentioned above, the decision was ultimately made to simply continue her immunosuppression with anakinra, hydroxychloroquine and high-dose steroids given her clinical improvement on that regimen alone.
By the time of follow-up 1 month later, this patient was also found to have the A91V monoallelic mutation in the PRF1 gene (also referred to as the FHLH2 gene). PRF1 encodes the protein perforin, which plays an essential role in the cytotoxic pathway. This mutation is actually not uncommon (for example, up to 17% in Caucasians [8]), and is debated as to whether it is pathogenic in heterozygous form [3, 8, 9] or even homozygous form [10]. A functional analysis of several genes involved in FHLH conducted by Trambas et al [8] and Voskoboinik et al [9] demonstrated that the A91V mutation reduces the stability and cytolytic activity of perforin, but without significant effect on overall perforin function. In the systematic review of Zhang et al, 150 patients were diagnosed with FHLH; they found that at least eight patients were heterozygous for the A91V mutation. Interestingly, they also found that one of the patients who was homozygous for A91V and died of HLH had a parent who was also homozygous but completely asymptomatic [10]. They concluded in their review that A91V was not pathogenic but rather a predisposing risk factor.
HLH/MAS continues to be a widely debated and investigated syndrome. There are various inclusion criteria for diagnosis, a growing literature of reported etiologies, and fascinating novel studies attempting to elucidate the genetics behind the syndrome. While FHLH and secondary HLH were previously separated into two separate disease processes, the growing database of genetic mutations found in patients with concurrent “secondary” etiologies is beginning to provide a case for the hypothesis that these triggers cause disease in patients who were already genetically predisposed [7, 9]. In this particular patient who ended up having an arguably predisposing genetic mutation, an active EBV infection, and AOSD, it was especially hard to retrospectively determine the driving source of her disease. However, it is important to emphasize that in the era of modern immunosuppressants, the use of etoposide and other chemotherapies is becoming more and more limited.
Acknowledgments
None to declare.
Financial Disclosure
None.
Conflict of Interest
None.
Informed Consent
Informed consent was obtained.
Author Contributions
JM and RG provided concept. RG prepared the manuscript. JM, RB and RG reviewed, edited and approved the final manuscript.
| References | ▴Top |
- Schulert GS, Canna SW. Convergent pathways of the hyperferritinemic syndromes. Int Immunol. 2018;30(5):195-203.
doi pubmed - Ravelli A, Davi S, Minoia F, Martini A, Cron RQ. Macrophage activation syndrome. Hematol Oncol Clin North Am. 2015;29(5):927-941.
doi pubmed - Gholam C, Grigoriadou S, Gilmour KC, Gaspar HB. Familial haemophagocytic lymphohistiocytosis: advances in the genetic basis, diagnosis and management. Clin Exp Immunol. 2011;163(3):271-283.
doi pubmed - Loh NK, Lucas M, Fernandez S, Prentice D. Successful treatment of macrophage activation syndrome complicating adult Still disease with anakinra. Intern Med J. 2012;42(12):1358-1362.
doi pubmed - Bergsten E, Horne A, Arico M, Astigarraga I, Egeler RM, Filipovich AH, Ishii E, et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study. Blood. 2017;130(25):2728-2738.
doi pubmed - Marsh RA. Epstein-Barr virus and hemophagocytic lymphohistiocytosis. Front Immunol. 2017;8:1902.
doi pubmed - Schram AM, Berliner N. How I treat hemophagocytic lymphohistiocytosis in the adult patient. Blood. 2015;125(19):2908-2914.
doi pubmed - Trambas C, Gallo F, Pende D, Marcenaro S, Moretta L, De Fusco C, Santoro A, et al. A single amino acid change, A91V, leads to conformational changes that can impair processing to the active form of perforin. Blood. 2005;106(3):932-937.
doi pubmed - Voskoboinik I, Thia MC, Trapani JA. A functional analysis of the putative polymorphisms A91V and N252S and 22 missense perforin mutations associated with familial hemophagocytic lymphohistiocytosis. Blood. 2005;105(12):4700-4706.
doi pubmed - Zhang K, Johnson JA, Biroschak J, Villanueva J, Lee SM, Bleesing JJ, Risma KA, et al. Familial haemophagocytic lymphohistiocytosis in patients who are heterozygous for the A91V perforin variation is often associated with other genetic defects. Int J Immunogenet. 2007;34(4):231-233.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.