

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://www.thejh.org |
Case Report
Volume 9, Number 3, September 2020, pages 84-88
Delayed Thrombotic Complications in a Thrombotic Thrombocytopenic Purpura Patient Treated With Caplacizumab
Nicolas Cillaa, Julie Dallemagneb, Marie Vanhovec, Patrick Stordeurd, Serge Mottee, Virginie De Wildeb, f
aDepartment of Internal Medicine, Faculty of Medicine, Universite Libre de Bruxelles, Brussels, Belgium
bHematology Department, Erasme Hospital, Brussels, Belgium
cEmergency Department, CHIREC Hospital, Braine-l’Alleud/Waterloo, Belgium
dImmunobiology Department, Erasme Hospital, Brussels, Belgium
eVascular Pathology Department, Erasme Hospital, Brussels, Belgium
fCorresponding Author: Virginie De Wilde, Clinic Hematology Department, Erasme Hospital, Route de Lennik, 808, 1070 Brussels, Belgium
Manuscript submitted February 17, 2020, accepted March 19, 2020, published online August 14, 2020
Short title: Delayed Thrombotic Complications in a TTP Patient
doi: https://doi.org/10.14740/jh614
| Abstract | ▴Top |
Thrombotic thrombocytopenic purpura (TTP) is a rare and unpredictable disease with a high mortality rate (90%) if untreated. It results from systemic microvascular thrombosis and leads to profound thrombocytopenia, hemolytic anemia and organ failure of varying severity. However, macrovascular thrombosis has been described in very rare cases. Caplacizumab has emerged as a promising new drug for the management of TTP. We report the case of a patient with idiopathic refractory TTP treated with caplacizumab who developed thrombotic complications upon discontinuation of treatment.
Keywords: Thrombotic thrombocytopenic purpura ; Refractory; Caplacizumab; Arterial thrombosis; Veinous thrombosis
| Introduction | ▴Top |
Thrombotic thrombocytopenic purpura (TTP) is a rare disease with an estimated annual incidence ranging from 1/250,000 to 1/1,000,000 [1]. It results from systemic microvascular thrombosis and leads to profound thrombocytopenia, hemolytic anemia and organ failure of varying severity. However, macrovascular thrombosis has been described in very rare cases [2]. Most cases of TTP in adults are caused by acquired auto-antibodies that inhibit ADAMTS13, a metalloprotease that cleaves von Willebrand factor (VWf), leading to VWf-platelet aggregation and microvascular thrombosis. TTP is a hematological emergency, with a mortality rate of 90% if untreated [3]. About 80% of patients respond to the first-line treatment which consists of corticosteroids and plasma exchange (PEX) to remove auto-antibodies and ultra-large VWf multimers and to replenish ADAMTS13 [4]. This treatment reduces the mortality to 10-15% [5]. In cases of TTP that are refractory to the first-line treatment, salvage therapies include twice-daily PEX, rituximab, cyclophosphamide, vincristine, cyclosporine and splenectomy [6]. Some experts recommend that rituximab should be introduced quickly in cases of failure of first-line treatment [6, 7]. However, the very low incidence of TTP is an obstacle to the development of clinical recommendations based on robust evidences and the very wide variability in survival rates across centers can be partly attributed to differences in management strategies due to insufficient guidance [8]. Recently, caplacizumab has emerged as a promising new drug for the management of TTP. This new drug is a nanobody that targets the A1 domain to the VWf and inhibits platelets adhesion to the VWf [9].
We report the case of a patient with idiopathic refractory TTP treated with caplacizumab who developed thrombotic complications upon discontinuation of treatment. To our knowledge, the occurrence of thrombotic complications with the discontinuation of caplacizumab has never been described.
| Case Report | ▴Top |
A 59-year-old man without any relevant clinical or surgical history presented to the emergency department on May 12, 2018, complaining of asthenia, dyspnea and palpitations for 5 days. He also had some red dots on his skin. His medical history included high blood pressure, hypercholesterolemia, gastroesophageal reflux disease and active smoking. He had no recent history of infection and no known allergies. His regular medications included pantoprazole, losartan, escitalopram and atorvastatin.
Upon physical examination, the patient’s clinical parameters were normal. Pallor, conjunctival jaundice and diffuse petechiae on the skin were noted. The rest of the clinical examination was normal. Blood tests revealed hemolytic normocytic and aregenerative anemia (Hb 7.2 g/dL) and severe thrombocytopenia (platelets count 13,000/mm3). A blood smear showed 35 - 40 schistocytes for every 1,000 red blood cells. The rest of the blood test was within normal limits. In particular, there was no renal insufficiency. Based on these results, TTP was the most likely diagnosis. The patient was transferred to the hematology department for appropriate medical care. The patient was promptly treated with daily PEX (1 × plasma volume) and methylprednisolone (1 mg/kg) in the intensive care unit (ICU).
The diagnosis of acquired TTP was confirmed by the presence of decreased ADAMTS13 activity to 0.5% (normal range 69-144%) and an anti-ADAMTS13 antibodies at 79 U/mL. An exhaustive etiologic investigation was inconclusive, including the positron emission tomography-computed tomography (PET-CT) and the anti-phospholipid serologies.
Initially, the patient’s platelet count rose to 89,000/mm3 but then dropped again to 29,000/mm3 after 7 days of treatment. Rituximab (375 mg/m2) was administered on days 1, 4, 8 and 15. At the same time, a daily high volume of plasma exchange (1.5 × plasma volume) and a bolus of solumedrol 1 g daily for 3 days were administered. Despite this intensive treatment, the patient developed a left sylvian stroke with right upper limb hemiparesis and Broca aphasia. The total recovery took 1 h. Magnetic resonance imaging (MRI) demonstrated multiple recent ischemic infra- and supratentorial lesions. The next day, the patient experienced NSTEMI acute coronary syndrome occurring with chest pain, impaired repolarization in V4, V5 and V6, and elevated troponin levels (401 ng/L). Cardiac ultrasound showed no alteration of kinetics and left ventricular ejection fraction. Acute renal failure also occurred with elevated creatinine at 1.4 mg/dL but this normalized within a few days. Treatment with vincristine (1.5 mg DT) was administered as the patient deteriorated. We also requested access to caplacizumab on day 13. Forty-eight hours after the introduction of caplacizumab, a rise in platelet count to 97,000/mm3 was noted and the disappearance of hemolysis stigmata (lactate dehydrogenase (LDH) 300 U/L and total bilirubin 1.7 mg/dL) was observed.
The patient was discharged on June 6, 2018, but was readmitted 5 days after discharge for non-thyphoid salmonella gastroenteritis complicated by bacteremia treated with ceftriaxone 2 g once daily for 7 days. The infection did not result in a relapse of TTP. A final course of rituximab was administered in view of residual B lymphocytosis and very slow normalization of ADAMTS13 activity (2 months). Caplacizumab was maintained until normalization of ADAMT13 levels (until August 9, 2018). Two weeks after stopping caplacizumab (August 23, 2018), the patient developed a deep vein thrombosis. Tinzaparin treatment was initiated for 3 months. Forty-eight hours after stopping tinzaparin (November 27, 2018), the patient developed severe intermittent claudication in the right arm.
A computed tomography angiography showed a thrombus in the brachiocephalic trunk, in the right common artery and in the distal right radial artery (Fig. 1). Treatment with acenocoumarol was then started and maintained permanently thereafter. Retrospectively, the patient reported some intermittent claudication in the right arm 4 months previously. It should also be noted that aspirin treatment was initiated just after the stroke and was maintained for a long period of time. The timeline summarizes the sequence of the patient's clinical events (Fig. 2).
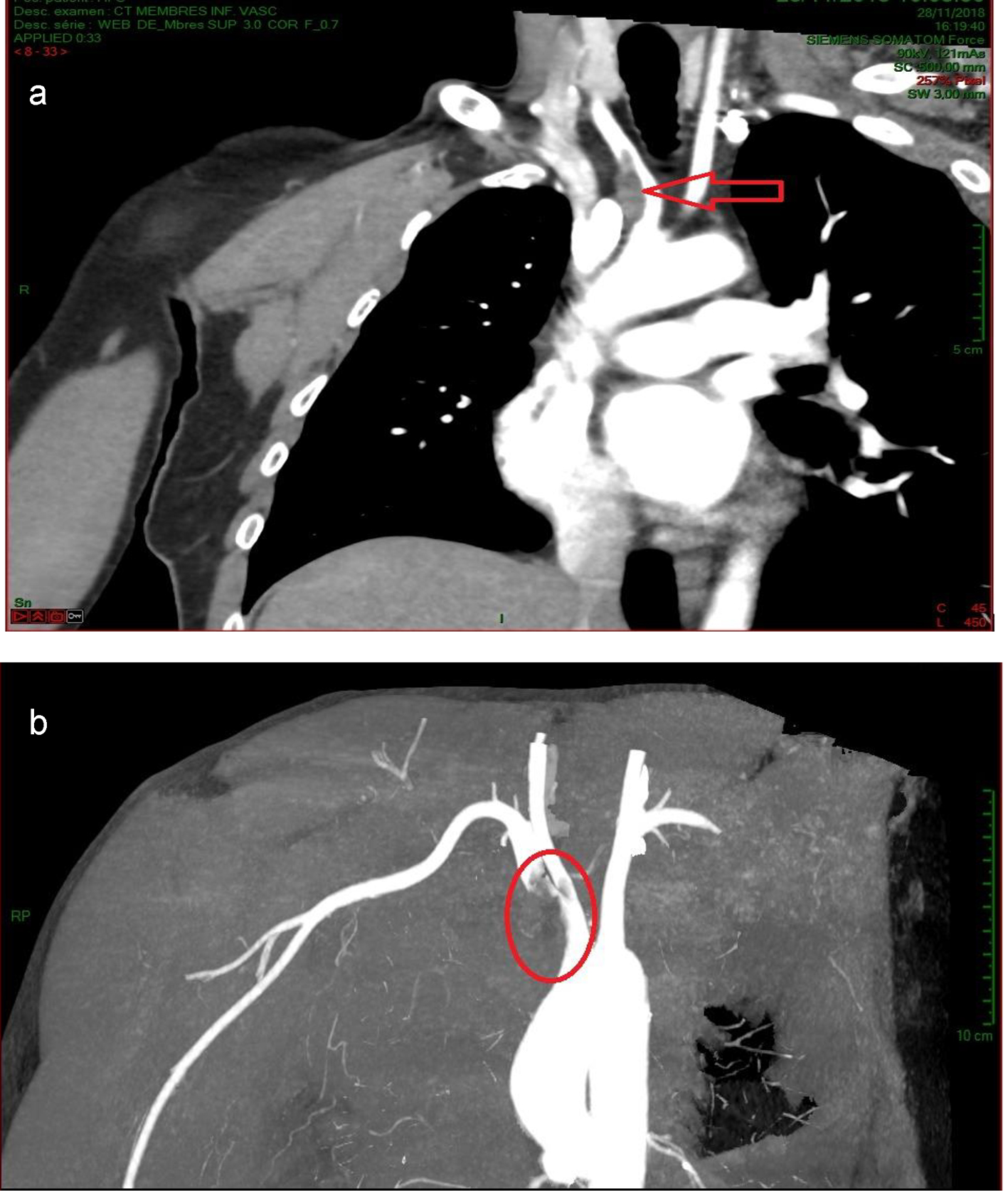 Click for large image | Figure 1. Computed tomography angiography showing a thrombus in the brachiocephalic trunk, in the right common artery (a) and in the distal right radial artery (b). |
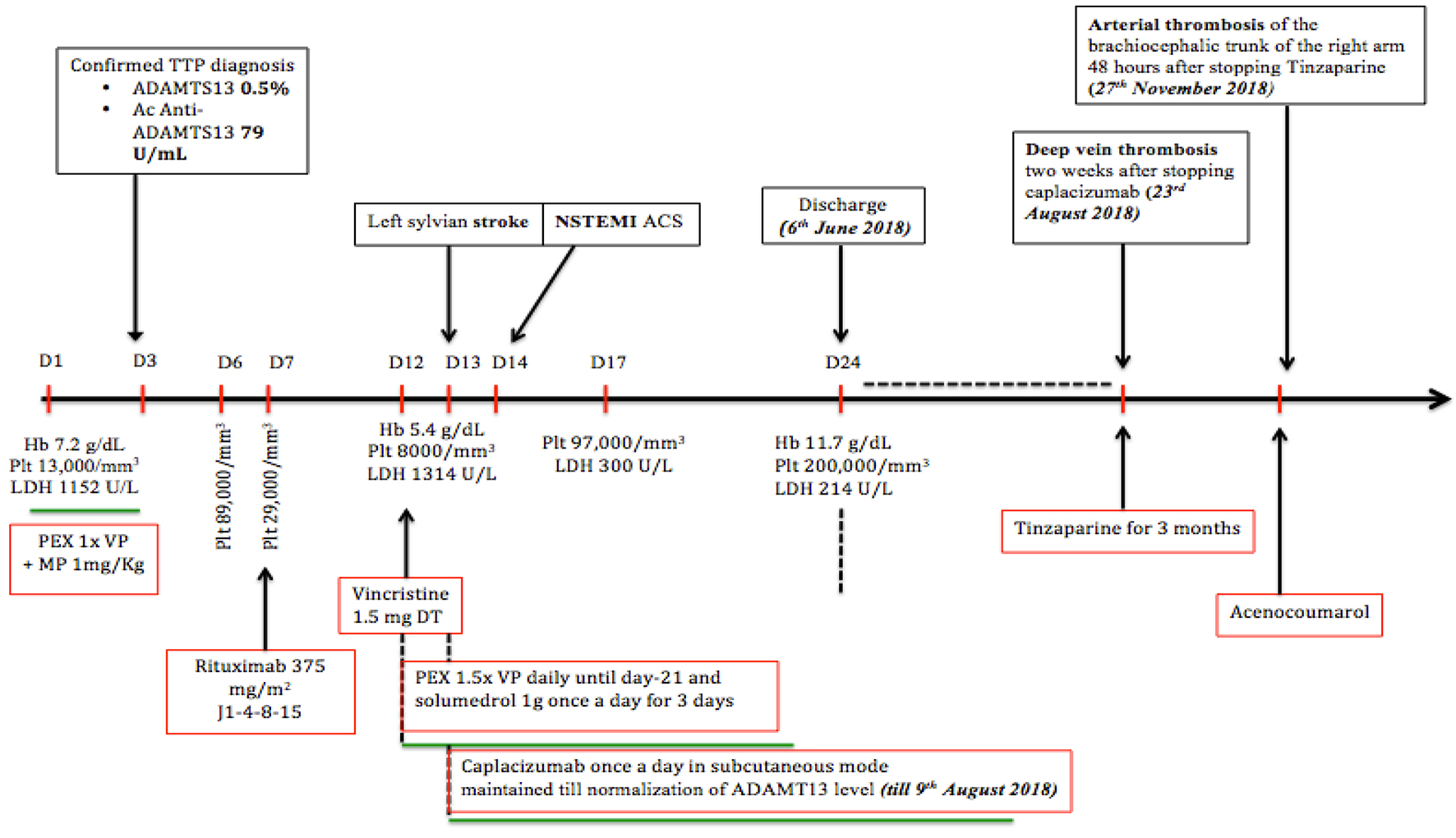 Click for large image | Figure 2. This timeline summarizes the patient’s clinical events. |
The patient had risk factors for atherothrombosis including arterial hypertension, hypercholesterolemia and active smoking. The readmission for non-typhoid salmonella bacteremia was an additional risk factor for thrombosis. There was no personal or family history of thrombosis. Thrombophilia assessment did not demonstrate any acquired predisposing factors (antiphospholipid syndrome, myeloproliferative syndromes with JAK2 mutation, PNH clone, or cancer). We also excluded genetic predisposing factors (antithrombin deficiency, C/S protein deficiency, resistance to activated protein C, prothrombin gene mutation).
| Discussion | ▴Top |
This case illustrates the unpredictability of this rare disease. Our patient was rapidly deteriorating and could not wait for the immunosuppressive effects of our treatment to come through. Through rapid blocking of VWf, caplacizumab prevents further platelet adhesion more rapidly than conventional treatment alone. Indeed, caplacizumab controls the formation of platelet aggregates until immunosuppressive treatments (particularly corticosteroids and rituximab), which have a slower onset of action, take effect. This opinion is also shared by Picod et al [10]. Two randomized controlled trials from 2016 and 2019 showed that caplacizumab induced a faster resolution of an acute TTP episode with a shorter time to normalization of platelet count [9, 11]. The study conducted by Scully et al also showed a very clinically significant reduction in deaths and recurrences of TTP [9]. With these results, some TTP experts recommend combining caplacizumab with first-line treatment (PEX and corticosteroids) [12].
Another important fact to note in this case is that our patient developed two thromboses (venous and arterial) when caplacizumab was stopped, while he was in complete hematological remission and his platelets were within the normal range. We know that microvascular thromboses are the prerogative of TTP. All organs may be involved, but some are more so than others. The nervous system is the first to be affected in 40-80% of cases. Renal (10-27%) and gastrointestinal (35-40%) involvements are also common. Cardiac damage may also occur in 25% of cases. The classic presentation combining hemolytic anemia, thrombocytopenia, fever, renal failure and neurological damage is only present in 7-10% of cases [6]. Macrovascular thrombosis has been described in very rare cases during the course of TTP [2]. However, the occurrence of macrovascular thrombosis in patients with TTP in complete hematological remission has not been described. Scully et al demonstrated that there were no differences in the venous thrombotic event rates between caplacizumab and placebo. Arterial events were rare post-treatment in both groups, and numbers were small, unless associated with an exacerbation or relapse of TTP [9]. The fact that our patient developed a venous thrombosis and then an arterial thrombosis after stopping the drug is surprising because caplacizumab, by inhibiting platelet adhesion to VWf, has bleeding side effects and, therefore, we could expect caplacizumab to have a protective effect on the occurrence of thrombosis. Did caplacizumab mask the thrombosis that was eventually revealed when the drug was stopped? Did the patient have a pro-thrombotic condition apart from TTP? Perhaps, but a complete thrombophilia check-up did not reveal anything. Yarranton et al recommend thromboprophylaxis with low-molecular-weight heparin once platelet count has reached > 50,000/mm3 [13]. The clinical efficacy of antiplatelet agents in TTP is unproven but they are relatively safe. Low dose aspirin (75 mg OD) may be given during platelet recovery (platelet count > 50,000/mm3) [14]. Is this thromboprophylaxis still necessary during caplacizumab treatment despite the risk of bleeding? As mentioned, the patient reported discomfort at the macrothrombosis sites while on caplacizumab.
Conclusions
This case highlights the advent of promising new therapies that may further improve the prognosis of this rare hematological disease. It also poses new questions regarding the management of TTP. We did not find any cases reporting the occurrence of thrombosis following the discontinuation of caplacizumab in the literature. This case raises the question of whether prophylaxis of thrombosis should be administered in association with caplacizumab. It also underlines the fact that we should be attentive to any subtle signs of thrombosis in patients treated with caplacizumab and to the risk of a potential flare of a thrombotic event at the withdrawal of the treatment.
Acknowledgments
None to declare.
Financial Disclosure
No funding was received. None of the authors have disclosures relevant to this manuscript.
Conflict of Interest
The authors declare no conflict of interest.
Informed Consent
The manuscript has been sufficiently de-identified to protect the patient.
Author Contributions
N. Cilla and J. Dallemagne co-wrote the article. M. Vanhove runs the emergency department where the diagnosis of TTP was made. P. Stordeur and S. Motte have been solicited several times for their expertise in thrombotic pathologies. V. De Wilde contributed to the reviewing of the article.
Data Avaibility
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Chiasakul T, Cuker A. Clinical and laboratory diagnosis of TTP: an integrated approach. Hematology Am Soc Hematol Educ Program. 2018;2018(1):530-538.
doi pubmed - Camous L, Veyradier A, Darmon M, Galicier L, Mariotte E, Canet E, Parquet N, et al. Macrovascular thrombosis in critically ill patients with thrombotic micro-angiopathies. Intern Emerg Med. 2014;9(3):267-272.
doi pubmed - Sadler JE. Pathophysiology of thrombotic thrombocytopenic purpura. Blood. 2017;130(10):1181-1188.
doi pubmed - Rock GA, Shumak KH, Buskard NA, Blanchette VS, Kelton JG, Nair RC, Spasoff RA. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian Apheresis Study Group. N Engl J Med. 1991;325(6):393-397.
doi pubmed - Sayani FA, Abrams CS. How I treat refractory thrombotic thrombocytopenic purpura. Blood. 2015;125(25):3860-3867.
doi pubmed - Joly BS, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura. Blood. 2017;129(21):2836-2846.
doi pubmed - Coppo P, Froissart A, French Reference Center for Thrombotic M. Treatment of thrombotic thrombocytopenic purpura beyond therapeutic plasma exchange. Hematology Am Soc Hematol Educ Program. 2015;2015:637-643.
doi pubmed - Azoulay E, Bauer PR, Mariotte E, Russell L, Knoebl P, Martin-Loeches I, Pene F, et al. Expert statement on the ICU management of patients with thrombotic thrombocytopenic purpura. Intensive Care Med. 2019;45(11):1518-1539.
doi pubmed - Scully M, Cataland SR, Peyvandi F, Coppo P, Knobl P, Kremer Hovinga JA, Metjian A, et al. Caplacizumab treatment for acquired thrombotic thrombocytopenic purpura. N Engl J Med. 2019;380(4):335-346.
doi pubmed - Picod A, Coppo P. Developments in the use of plasma exchange and adjunctive therapies to treat immune-mediated thrombotic thrombocytopenic purpura. Expert Rev Hematol. 2019;12(6):461-471.
doi pubmed - Peyvandi F, Scully M, Kremer Hovinga JA, Cataland S, Knobl P, Wu H, Artoni A, et al. Caplacizumab for acquired thrombotic thrombocytopenic purpura. N Engl J Med. 2016;374(6):511-522.
doi pubmed - Knoebl P, Cataland S, Peyvandi F, Coppo P, Scully M, Kremer Hovinga JA, Metjian A, et al. Efficacy and safety of open-label caplacizumab in patients with exacerbations of acquired thrombotic thrombocytopenic purpura in the HERCULES study. J Thromb Haemost. 2020;18(2):479-484.
doi pubmed - Yarranton H, Cohen H, Pavord SR, Benjamin S, Hagger D, Machin SJ. Venous thromboembolism associated with the management of acute thrombotic thrombocytopenic purpura. Br J Haematol. 2003;121(5):778-785.
doi pubmed - Bobbio-Pallavicini E, Gugliotta L, Centurioni R, Porta C, Vianelli N, Billio A, Tacconi F, et al. Antiplatelet agents in thrombotic thrombocytopenic purpura (TTP). Results of a randomized multicenter trial by the Italian Cooperative Group for TTP. Haematologica. 1997;82(4):429-435.
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.