

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://www.thejh.org |
Case Report
Volume 10, Number 3, June 2021, pages 130-135

A Case Series of TERC Variant Telomere Biology Disorders in Unrelated Families From Atlantic Canada
Amye M. Harrigana ![]() , Shelley MacDonaldb, Bruce Crooksc, Sarah Dyackb, d, Amy M. Trottiera, e, f
, Shelley MacDonaldb, Bruce Crooksc, Sarah Dyackb, d, Amy M. Trottiera, e, f ![]()
aDepartment of Medicine, Queen Elizabeth II Health Science Centre, Halifax, NS, Canada
bMaritime Medical Genetics Service, IWK Health, Halifax, NS, Canada
cDivision of Pediatric Hematology and Oncology, IWK Health, Halifax, NS, Canada
dDivision of Pediatric Medical Genetics, IWK Health, Halifax, NS, Canada
eDivision of Hematology, Queen Elizabeth II Health Science Centre, Halifax, NS, Canada
fCorresponding Author: Amy M. Trottier, Division of Hematology, Nova Scotia Health Authority/Dalhousie University, Room 427, Bethune Building, 1276 South Park Street, Halifax, NS B3H 2Y9, Canada
Manuscript submitted March 26, 2021, accepted April 27, 2021, published online June 16, 2021
Short title: TERC Variant TBDs in Atlantic Canada
doi: https://doi.org/10.14740/jh826
| Abstract | ▴Top |
TERC variant telomere biology disorders (TBDs) are a rare, heterogenous group of disorders that arise from germline variants in TERC, a gene that encodes for the RNA component of telomerase. Variants in TERC lead to accelerated telomere attrition and can manifest as many different phenotypes. In this case series, we aimed to add to the literature describing TERC variant TBDs by reporting cases from two unrelated families from Atlantic Canada. The first case, a previously described germline TERC variant, n.107G>T (NR_001566.1), was identified in a young woman with myelodysplastic syndrome (MDS) and found to segregate with cytopenias in the family. This case represents a unique phenotypic presentation: this variant has not previously been described in patients with MDS and adds important segregation data to the literature. The second case, a novel TERC n.437T>G variant, was identified in a patient with both aplastic anemia and pulmonary fibrosis manifesting in his early 30s. We report these novel cases of germline TERC variants in order to help clinicians recognize TBDs, as well as to add important supporting information for the pathogenicity of these variants.
Keywords: Telomere biology disorder; TERC; Myelodysplastic syndrome; Pulmonary fibrosis; Aplastic anemia
| Introduction | ▴Top |
Telomere biology disorders (TBDs) are a heterogenous group of rare disorders that arise from germline variants in genes that encode components of telomerase and/or telomere maintenance genes [1]. Variants in these genes can lead to significantly shortened telomere lengths and can impact multiple organ systems [1]. Advances in genetic sequencing and its increased use in clinical medicine have helped expand our awareness and understanding of TBDs; however, they are frequently unrecognized and underdiagnosed. Of the numerous genes that have been identified as being implicated in TBDs, TERC is one of the most frequently reported [2]. TERC encodes the RNA component of telomerase, an important DNA polymerase that helps maintain the integrity of telomeres [1]. Pathogenic TERC variants are inherited in an autosomal dominant manner and have been found to result in short telomere length, reduced telomerase activity and accelerated telomere attrition [3].
To date, there are over 60 TERC variants reported to be associated with TBDs [2]. Given the heterogenous presentations of TERC variant TBDs, it is important to report novel TERC variants or/and novel phenotypes of previously reported TERC variants to help clinicians recognize and diagnose these TBDs. Making this diagnosis carries important implications for the management of patients and their families. We aimed to add to the literature of TERC variant TBDs by describing two unrelated families from Atlantic Canada with two different germline TERC variants, one of which is a novel variant and the other a novel phenotype of a previously reported TERC variant.
| Case Reports | ▴Top |
Family A
Proband A (III-1), a 33-year-old Caucasian female of non-Finnish European descent, was referred to our center’s inherited hematologic malignancy clinic. She originally presented with severe thrombocytopenia and macrocytic anemia at age 19, while pregnant. During the pregnancy, her platelets (PLT) and hemoglobin (HGB) progressively declined, reaching a nadir of 38 × 109/L and 80 g/L, respectively, at 33 weeks gestational age. A bone marrow aspiration and biopsy were performed at the end of the second trimester of pregnancy and revealed multilineage dysplasia, 1% blasts and normal cytogenetics. She had no other issues throughout the pregnancy and delivered a healthy girl (IV-1) via spontaneous vaginal delivery at 37 + 5 weeks gestational age. Although HGB and PLT count improved post-partum, she continued to have a mild macrocytic anemia and thrombocytopenia. A second bone marrow aspiration and biopsy done post-partum showed ongoing multilineage dysplasia with 1% blasts and normal cytogenetics. She was diagnosed with low risk myelodysplastic syndrome (MDS) with a Revised International Prognostic System (R-IPSS) score of 2.5. She has a very rare human leukocyte antigen (HLA) type with no available sibling matches (Fig. 1).
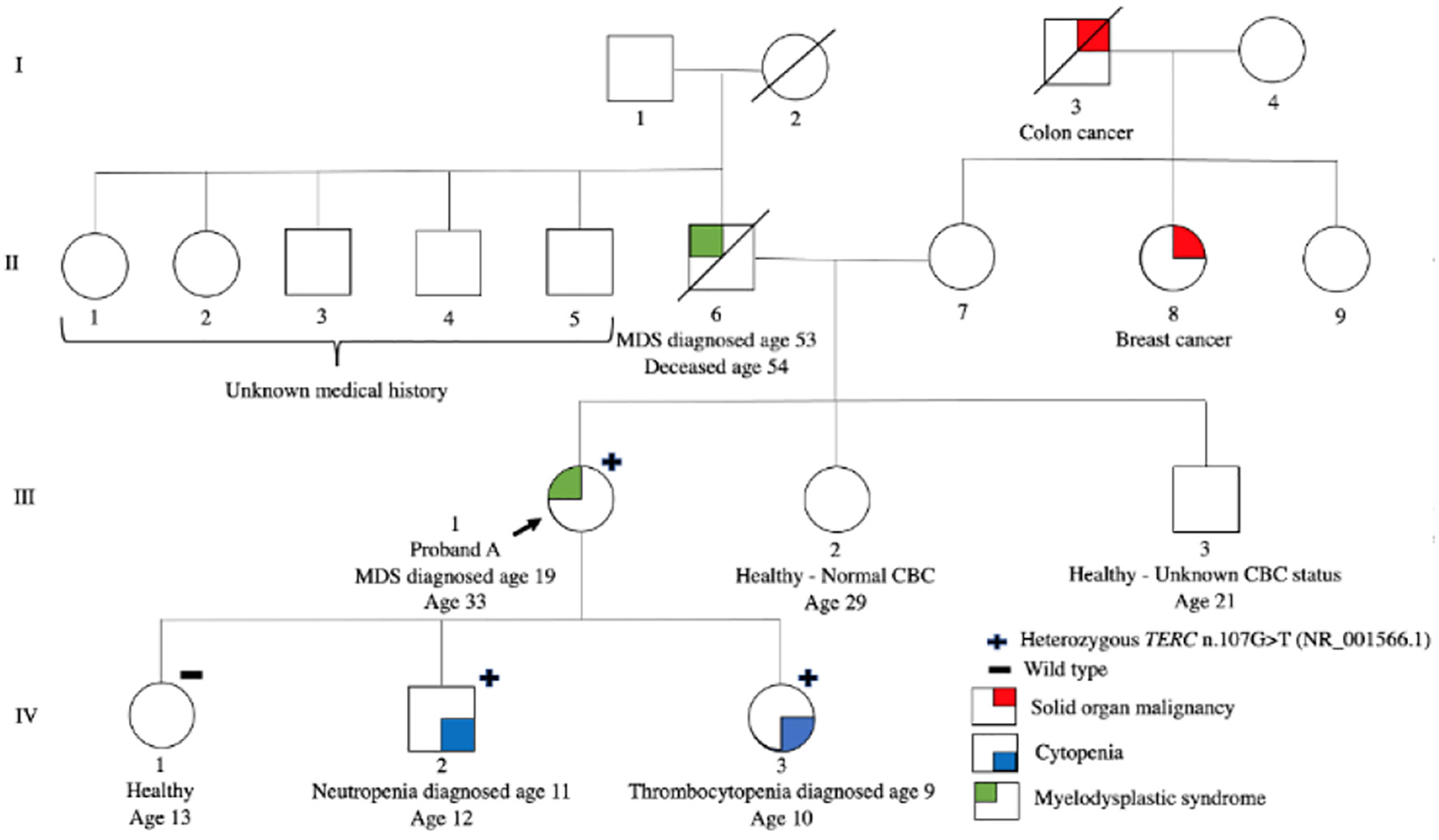 Click for large image | Figure 1. Pedigree of family A. Roman numerals indicate generations and arabic numbers indicate individuals. The proband is indicated by an arrow. The individuals who are heterozygous for the TERC n.107G>T are marked with a +. Individual(s) who were tested for TERC n.107G>T and did not have the variant (wild type) are marked with a -. Phenotypes are represented by the following colors: red signifies solid organ malignancy, blue signifies cytopenia and green signifies myelodysplastic syndrome. |
Proband A had two additional pregnancies, with the last being complicated by a premature rupture of membranes at 29 weeks’ gestation and preterm delivery of a girl (IV-3) at 31 weeks’ gestation via cesarean section. Proband A’s past medical history was also significant for premature graying of hair as a teenager. Proband A has remained asymptomatic and transfusion independent with stable moderate thrombocytopenia and macrocytosis for the subsequent 13 years without the need for any disease-directed therapies.
Proband A’s father (II-6) was diagnosed with MDS at the age of 53 years and he died within a year of diagnosis. There was no history of hematologic malignancy nor hematologic abnormalities on the maternal side of the family. Proband A has two siblings, one of whom is healthy and had a normal complete blood cell count (CBC) (III-2), but the hematologic status of her other sibling is unknown (III-3).
Given the significant family history, proband A’s three children were referred to pediatric hematology for evaluation. The two youngest children, one male and one female, were found to have leukopenia and thrombocytopenia as well as macrocytosis at ages 11 years (IV-2) and 9 years old (IV-3), respectively (Table 1). The male (IV-2) was otherwise healthy with normal physical examination. The youngest daughter (IV-3) was born prematurely, had a history of easy bruising and had scattered ecchymosis and numerous dental caries on examination. The eldest (IV-1), a 12-year-old girl, was healthy, with normal physical examination, and no CBC abnormalities.
 Click to view | Table 1. Summary of Phenotypic Characteristics and Relevant Diagnostic Investigations in the Individuals From Families A and B |
Germline genetic testing using a 41-gene Hereditary Leukemia Next Generation Sequencing (NGS) Panel (BluePrint Genetics) was conducted for proband A using DNA extracted from cultured skin fibroblasts and revealed a heterozygous germline TERC n.107G>T (NR_001566.1) variant. Tumor-based testing from peripheral blood using the Ilumina TruSight Myeloid panel (a 54-gene NGS panel) identified a somatic missense variant in a splicing factor gene (U2AF1 c.101C>T, pS34F). Notably, TERC is not included within the TruSight Myeloid panel and hence this variant was not detected in the patient’s blood sample. Telomere length (TL) was less than the first percentile for age for total lymphocytes and all lymphocyte subsets. Proband A’s three children underwent targeted familial variant testing for the germline TERC variant, and the two youngest children were found to be heterozygous for the same variant. The eldest child, with normal CBC, did not inherit the variant and had normal TL whereas the son and younger daughter both had TL less than the first percentile for age for total lymphocytes and all lymphocyte subsets. The son’s bone marrow aspiration and biopsy showed a normocellular marrow with no dysplasia, no increase in blasts and normal cytogenetics and the younger daughter’s bone marrow aspiration and biopsy showed rare dysplastic erythroblasts and megakaryocytes not meeting criteria for MDS (Table 1). HLA typing of all three children and the proband confirmed shared maternity and paternity status.
Family B
Proband B (III-2), a male of mixed French (maternal) and Scandinavian descent (paternal), presented at age 35 years with complaints of generalized fatigue and shortness of breath. Investigations revealed a mild macrocytic anemia (HGB 124 g/L (mean corpuscular volume (MCV) 104.5)), thrombocytopenia (PLT 95 × 109/L) and a normal neutrophil count. Bone marrow biopsy and aspiration showed a markedly hypocellular marrow for age (20-30% cellularity), consistent with non-severe aplastic anemia (AA) (Table 1). Computed tomography scan was consistent with diagnosis of pulmonary fibrosis (PF) and pirfenidone was started. His past medical history was notable for childhood asthma managed with bronchodilators as needed, premature graying of the hair starting at age 19, type 2 diabetes managed with oral anti-hyperglycemics, obesity and a cholecystectomy. He smoked approximately half a pack of cigarettes daily. Further investigations revealed normal mitomycin C and diepoxybutane chromosomal breakage tests and no evidence of a paroxysmal nocturnal HGB clone; however, telomere lengths were markedly abnormal, measuring less than the first percentile for age for total lymphocytes and all lymphocyte subsets, suggestive of an underlying TBD (Fig. 2).
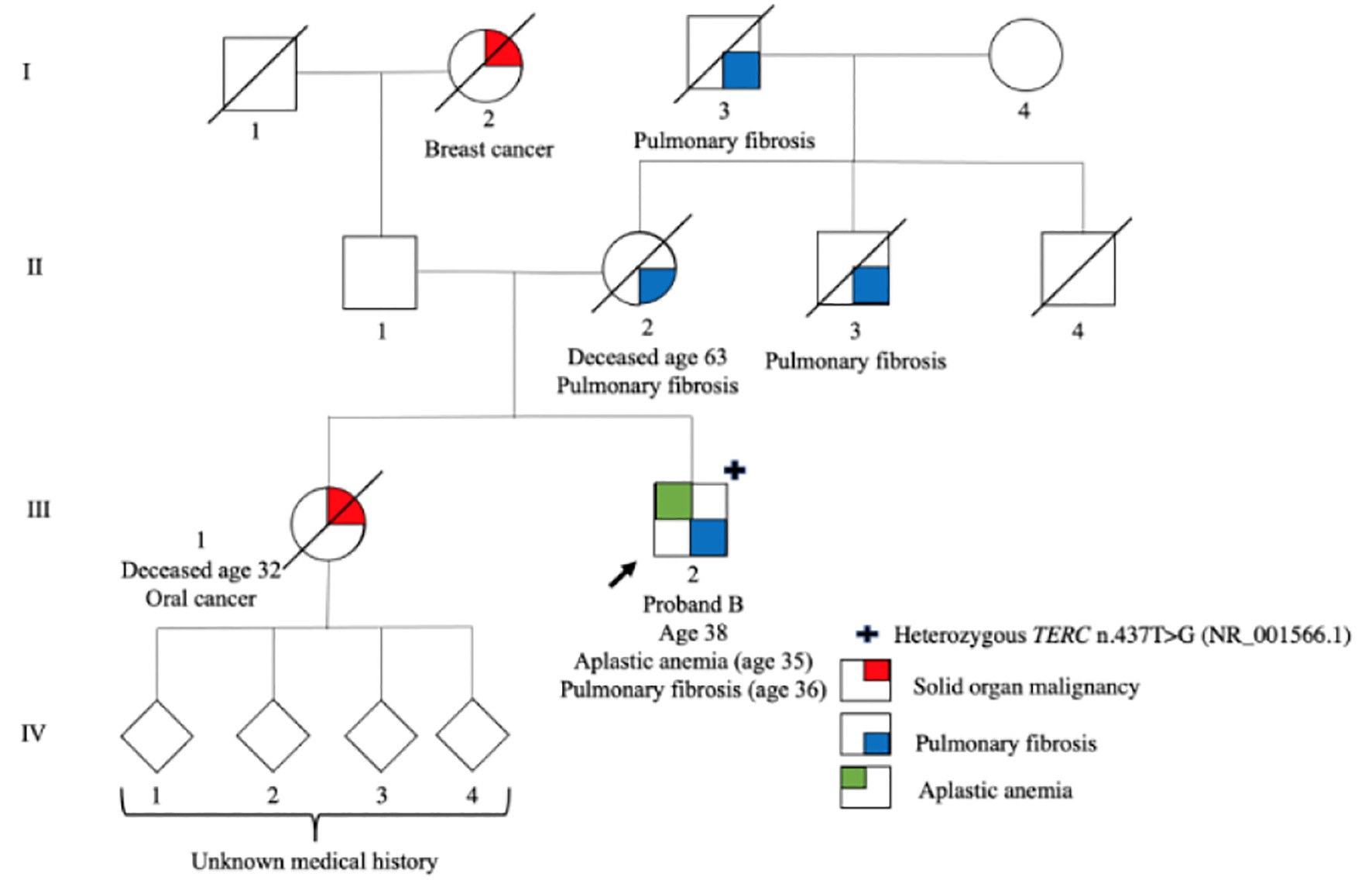 Click for large image | Figure 2. Pedigree of family B. Roman numerals (I-IV) indicate generations and arabic numbers indicate individuals. The proband is indicated by an arrow. The individual(s) who are heterozygous for the TERC n.437T>G are marked with a +. Phenotypes are represented by the following colors: red signifies solid organ malignancy, blue signifies pulmonary fibrosis and green signifies aplastic anemia. |
Proband B’s family history was significant for PF and solid tumors. His mother (II-2) and maternal uncle (II-3) both died from complications of PF. His maternal grandfather (I-3), who worked as a coalminer, died from pulmonary disease presumed to be coal worker’s pneumonoconiosis, however, in retrospect, was suspicious for PF. Proband B’s sister (III-1), who was also a smoker, died from oral cancer at age 32. She had four children (IV-1-4), but their medical history and health status is unknown. The family history of the proband’s paternal side was not well known; there was apparently no history of PF or hematologic abnormalities. His paternal grandmother (I-2) had breast cancer and died at an unknown age.
Proband B’s genetic testing performed from peripheral blood using the Blueprint Genetics Bone Marrow Failure Syndrome NGS panel, a 135-gene panel, revealed heterozygosity for TERC n.437T>G (NR_001566.1). Because the NGS was performed on a peripheral blood sample, the germline nature of this variant cannot be concluded with certainty. However, given the gene involved, the variant’s presence at a variant allele frequency (VAF) of about 50%, and the strong personal and family history, all in keeping with a TBD, we suspect the variant to be germline. As other members of the family with clinical features of TBDs were deceased, segregation studies could not be conducted.
| Discussion | ▴Top |
Many different phenotypes associated with TERC variants have been described in the literature [2, 4]. In our cases, both families demonstrated phenotypes consistent with previously reported TERC variants; family A’s variant (TERC n.107G>T) manifested as MDS and cytopenia(s) that exhibited genetic anticipation within successive generations. Additional features present that can be consistent with TBDs included premature greying of hair, preterm birth and dental caries [5-7]. Family B’s TERC variant (TERC n.437T>G) manifested as AA and PF in the proband with a significant family history of PF and a case of head and neck cancer at an early age, all of which are consistent with TERC variant TBDs [8-11].
Genetic anticipation, the phenomenon of earlier disease onset and increased severity of disease with each successive generation, is a postulated feature of TERC TBDs [1, 12]. The mechanism by which genetic anticipation occurs in TBDs is hypothesized to be two-fold: 1) significantly shorter telomeres present in germ cells are passed down to the progeny; and 2) the progeny also inherits the telomerase gene variant, which leads to further telomere attrition over time [1, 12]. This concept is illustrated in our case series, particularly by the proband of family B, who developed bone marrow failure in addition to PF.
The TERC n.107G>T variant found in family A has previously been reported in two cases: 1) a 29-year-old patient with constitutional AA, microcephaly and aseptic necrosis of the femoral head [13, 14]; and 2) a 64-year-old female with anemia, PF and multiple malignancies [14, 15]. To the best of our knowledge, this is the first report of TERC n.107G>T manifesting as MDS as well as the first report to demonstrate segregation of this variant with hematologic abnormalities including MDS and unexplained cytopenias. Prior to our case, the TERC n.107G>T variant would have been classified as likely pathogenic (PS3, PM1, PM2, PP4), based on the American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) criteria [16]. With the incorporation of the segregation data from our case, this variant can now be classified as pathogenic (PS3, PM1, PM2, PP1 and PP4) [16].
The TERC n.437T>G variant detected in the proband B is not found in the gnomAD database nor has it previously been described in the literature. According to the ACMG/AMP criteria, this variant is classified as a variant of uncertain significance (VUS) (PM1, PM2 and PP4) [16]. Although this variant is currently a VUS, because of the clinical diagnosis of a short telomere syndrome in proband B, and the strong maternal family history consistent with a short telomere syndrome, we suspect this genetic change is a true pathogenic variant.
Learning points
Our cases add important new information to the body of literature describing the heterogeneous nature of TERC variant TBDs. We described a new phenotypic presentation for the TERC n.107G>T variant and add valuable evidence for segregation. With this additional evidence, this variant can now be classified as pathogenic according to the ACMP/AMP criteria. We also described a novel variant, TERC n.437T>G, and its associated phenotype of AA and PF. Our cases highlight the heterogeneity and complexity of TERC TBDs and the need for increased awareness of these disorders. When a TBD is suspected, individuals and their families should be referred to appropriate specialists to help ensure a timely diagnosis. The diagnostic algorithm proposed by Townlsey et al (2014) is a helpful resource that can guide clinicians through the initial steps in the diagnosis of TBDs [3]. Once a diagnosis is established, affected individuals will require ongoing surveillance for the prevention and early detection of the multiorgan system complications of TBDs.
Acknowledgments
None to declare.
Financial Disclosure
This research was supported by funds from the QEII Health Sciences Centre Foundation.
Conflict of Interest
The authors of this case series do not have any conflict of interest to disclose.
Informed Consent
As per our institution’s Research and Ethics Board policy, written informed consent was obtained from the patients and legal guardians of patients whose personal medical information was included in this case series prior to submission of the manuscript. The consent forms were signed electronically given the COVID-19 pandemic restrictions.
Author Contributions
AT identified the cases and obtained written informed consent for the case series as well as supervised and revised the manuscript. AH wrote the manuscript and participated in the revisions of the manuscript. SM, SD and BC were involved in the care of the patients featured in the case series. All authors edited, read, and approved the final manuscript.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
Abbreviations
TBD: telomere biology disorder; MDS: myelodysplastic syndrome; PLT: platelets; HGB: hemoglobin; CBC: complete blood count; NGS: next generation sequencing; TL: telomere length; AA: aplastic anemia; PF: pulmonary fibrosis; VAF: variant allele frequency; VUS: variant of uncertain significance
| References | ▴Top |
- Armanios M, Blackburn EH. The telomere syndromes. Nat Rev Genet. 2012;13(10):693-704.
doi pubmed - Nagpal N, Agarwal S. Telomerase RNA processing: Implications for human health and disease. Stem Cells. 2020;38(12):1532-1543.
doi pubmed - Townsley DM, Dumitriu B, Young NS. Bone marrow failure and the telomeropathies. Blood. 2014;124(18):2775-2783.
doi pubmed - University of Chicago Hematopoietic Malignancies Cancer Risk Team. How I diagnose and manage individuals at risk for inherited myeloid malignancies. Blood. 2016;128(14):1800-1813.
doi pubmed - Gansner JM, Achebe MM, Gray KJ, Yefidoff-Freedman R, Labovitis E, Parnes A, Connors JM, et al. Pregnancy outcomes in inherited bone marrow failure syndromes. Blood. 2017;130(14):1671-1674.
doi pubmed - Atkinson JC, Harvey KE, Domingo DL, Trujillo MI, Guadagnini JP, Gollins S, Giri N, et al. Oral and dental phenotype of dyskeratosis congenita. Oral Dis. 2008;14(5):419-427.
doi pubmed - Vulliamy TJ, Marrone A, Knight SW, Walne A, Mason PJ, Dokal I. Mutations in dyskeratosis congenita: their impact on telomere length and the diversity of clinical presentation. Blood. 2006;107(7):2680-2685.
doi pubmed - Newton CA, Batra K, Torrealba J, Kozlitina J, Glazer CS, Aravena C, Meyer K, et al. Telomere-related lung fibrosis is diagnostically heterogeneous but uniformly progressive. Eur Respir J. 2016;48(6):1710-1720.
doi pubmed - Borie R, Tabeze L, Thabut G, Nunes H, Cottin V, Marchand-Adam S, Prevot G, et al. Prevalence and characteristics of TERT and TERC mutations in suspected genetic pulmonary fibrosis. Eur Respir J. 2016;48(6):1721-1731.
doi pubmed - Armanios MY, Chen JJ, Cogan JD, Alder JK, Ingersoll RG, Markin C, Lawson WE, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356(13):1317-1326.
doi pubmed - Parry EM, Alder JK, Qi X, Chen JJ, Armanios M. Syndrome complex of bone marrow failure and pulmonary fibrosis predicts germline defects in telomerase. Blood. 2011;117(21):5607-5611.
doi pubmed - Vulliamy T, Marrone A, Szydlo R, Walne A, Mason PJ, Dokal I. Disease anticipation is associated with progressive telomere shortening in families with dyskeratosis congenita due to mutations in TERC. Nat Genet. 2004;36(5):447-449.
doi pubmed - Vulliamy TJ, Kirwan MJ, Beswick R, Hossain U, Baqai C, Ratcliffe A, Marsh J, et al. Differences in disease severity but similar telomere lengths in genetic subgroups of patients with telomerase and shelterin mutations. PLoS One. 2011;6(9):e24383.
doi pubmed - National Centre for Biotechnology Information. TERC [gene]. 2020. https://www.ncbi.nlm.nih.gov/clinvar (Accessed May 19, 2020).
- Norberg A, Rosen A, Raaschou-Jensen K, Kjeldsen L, Moilanen JS, Paulsson-Karlsson Y, Baliakas P, et al. Novel variants in Nordic patients referred for genetic testing of telomere-related disorders. Eur J Hum Genet. 2018;26(6):858-867.
doi pubmed - Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-424.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.