

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://www.thejh.org |
Letter to the Editor
Volume 10, Number 6, December 2021, pages 274-276

Myelodysplastic Syndrome Associated With Auto-Immune Inflammatory Disease in VEXAS Syndrome
Vibha Taneja Thomasa, c ![]() , Mohan Penmetchab
, Mohan Penmetchab
aDepartment of Medical Oncology, Texas Oncology PA, Dallas, TX, USA
bDepartment of Rheumatology, Texas Health Presbyterian Hospital, Plano, TX, USA
cCorresponding Author: Vibha Taneja Thomas, Department of Medical Oncology, Texas Oncology, 12221 Merit Dr. #500, Dallas, TX 75251, USA
Manuscript submitted October 27, 2021, accepted December 7, 2021, published online December 21, 2021
Short title: Myelodysplastic Syndrome
doi: https://doi.org/10.14740/jh940
| To the Editor | ▴Top |
Recently, VEXAS (vacuoles, E1 enzyme, X-linked, auto-inflammatory, somatic) syndrome has surfaced as a new disease entity linking an auto-inflammatory clinical presentation with an underlying hematologic disorder. Recently published case reports outline the clinical course and hematologic pathologies associated with this condition.
We present a 62-year-old male, with history of type 2 diabetes mellitus and coronary artery disease, who presented with a 6-month history of intermittent fevers, fatigue, body aches, weight loss of 30 lbs, and palpable purpuric lesions on the lower extremities. His medications included metoprolol, ranolazine, prasugrel, atorvastatin, alirocumab, insulin, liraglutide, empagliflozin and cyanocobalamin. Physical examination was unremarkable. In particular there was no clinical evidence of inflammatory arthritis or muscle weakness. Blood tests were significant for low hemoglobin of 10.1 g/dL, low hematocrit of 30%, elevated mean corpuscular volume of 108.3 fL, elevated erythrocyte sedimentation (ESR) rate of 97 mm/h and elevated C-reactive protein (CRP) of 6.9 mg/dL. Total white blood cell count (WBC) was 5,100 cells/µL with normal differential and platelet count was 176,000/µL. Rheumatoid factor, antinuclear antibodies, antineutrophil cytoplasmic antibodies and cryoglobulins were negative. Serum protein electro-phoresis (SPEP) was negative for M-protein. B12 and folic acid levels were normal. Ferritin was elevated at 413 ng/mL, consistent with noted inflammatory state. No proteinuria was identified. Skin biopsy of the thigh lesion showed nuclear dust in the superficial dermis and scattered neutrophilic infiltration and fibrinoid necrosis of the blood vessels consistent with leukocytoclastic vasculitis. Computed tomography of the lungs showed bilateral nodules measuring up to 9 mm. Some nodules were new and others had resolved when compared to previous computed tomography done in 2010. They were described as indeterminate and may be infectious, inflammatory, or neoplastic. Bone marrow aspirate findings were consistent with myelodysplastic syndrome with multilineage dysplasia. Marrow was noted to be hypercellular with greater than 95% cellularity with trilineage dysplasia (Fig. 1) and 12% ring sideroblasts. No increase in blasts was noted. A portion of erythroid and myeloid precursors showed cytoplasmic vacuolization as well as hypogranular neutrophils with nuclear projections (Fig. 2). Cytogenetics revealed normal male karyotype. Fluorescent in situ hybridization (FISH) probes for 5q31, 5p15.2, 7q31, 7 centromere, 8 centromere, 11q23 and 20q12 were all normal. Next generation sequencing for myelodysplastic syndrome (MDS) with assessment of 38 genes (ASXL1, ATM, BCOR, CBL, CEBPA, CSF3R, CUX1, DNMT3A, ETNK1, ETV6, EZH2, FLT3, GATA2, IDH1, IDH2, IKZF1, JAK2, KRAS, NF1, NPM1, NRAS, PHF6, PTEN, PTPN11, RUNX1, SETBP1, SF3B1, SRSF2, STAG2, STAT3, STK11, TET2, TP53, U2AF1, WT1, and ZRSR2) was also negative for pathogenic mutations. International Prognostic Scoring System (IPSS) risk score was calculated as low risk with score of 0. Our patient’s medication history did not support medication-induced vacuolization of hematopoietic stem cells.
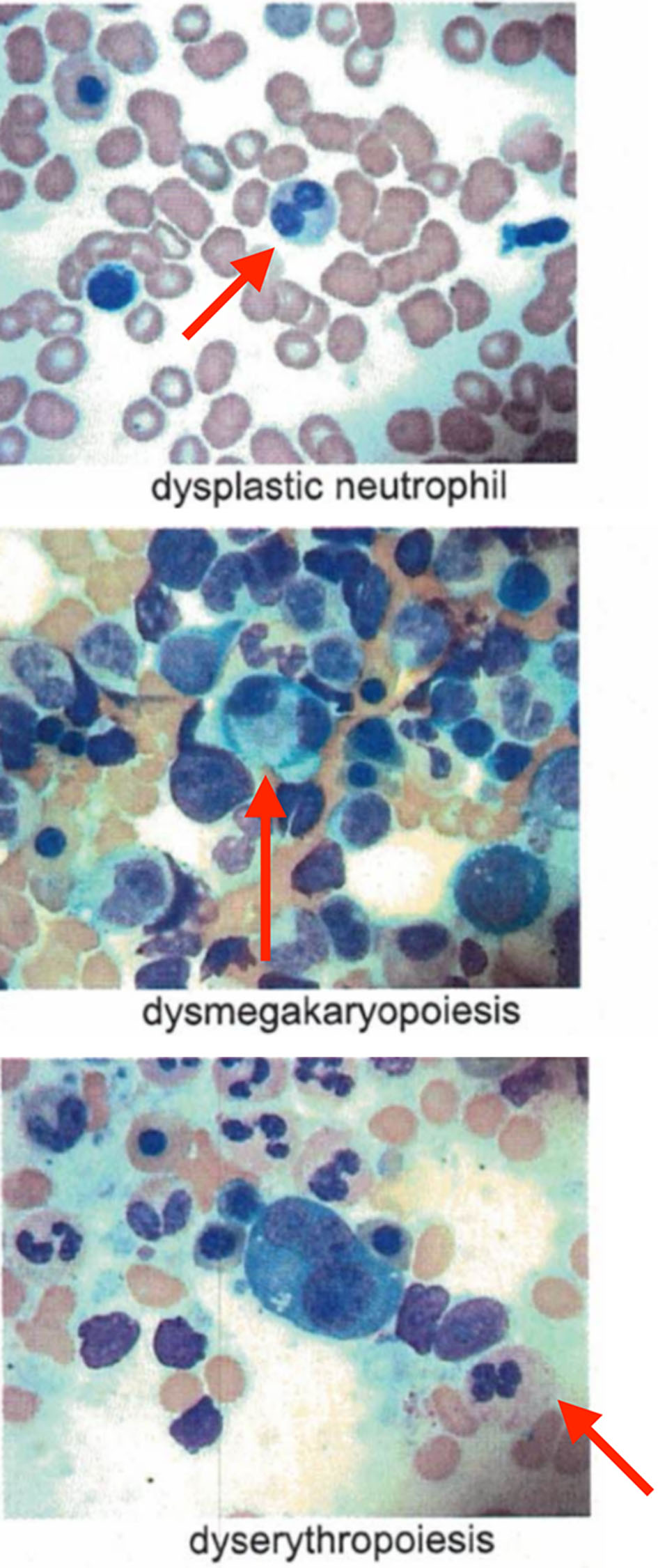 Click for large image | Figure 1. Trilineage dysplasia noted in bone marrow aspirate. |
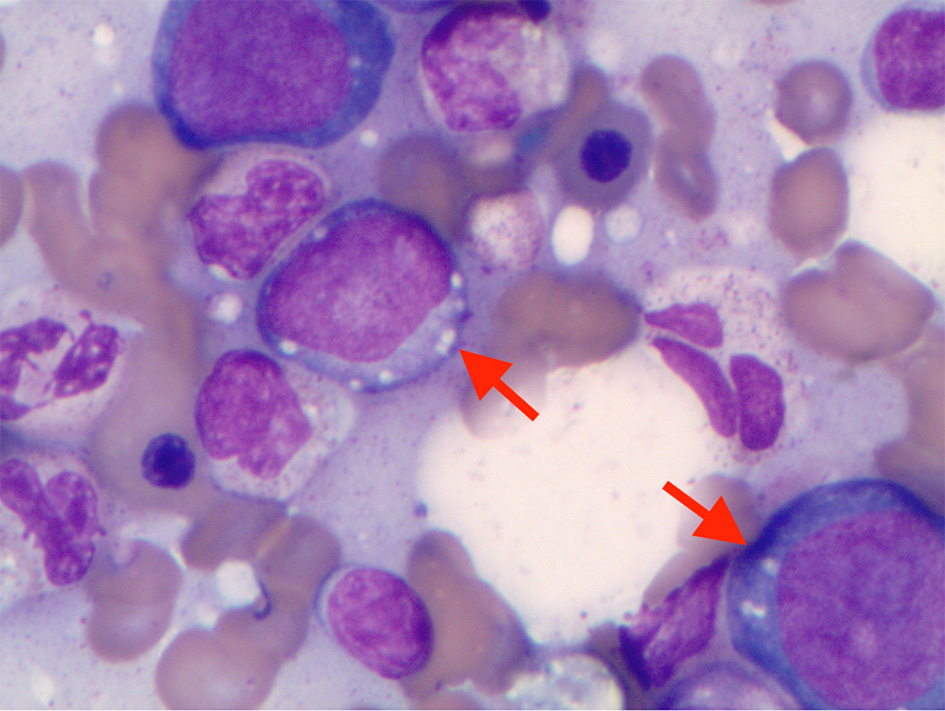 Click for large image | Figure 2. Cytoplasmic vacuoles in erythroid and myeloid precursors in bone marrow aspirate. |
Given the conglomerate of noted inflammatory, radiographic, and hematologic findings, genetic testing was ordered. Sequence analysis and deletion/duplication testing for the UBA1 gene in this patient revealed UBA1, c.121A>C (p.Met41Leu) variant. Sequence data suggested possible mosaicism in this sample (variant present in some cells and absent from others) with variant allele frequency of 57-67%. No other variants in the UBA1 gene were identified. This patient’s clinical presentation along with bone marrow aspirate findings and evidence of possible mosaic UBA1 gene mutation suggested a diagnosis of VEXAS syndrome.
VEXAS syndrome is a newly described auto-inflammatory condition affecting older adults. Somatic mutations are identified affecting methionine-41 (p.Met41) in the UBA1 gene on the X-chromosome of myeloid cells that encodes the E1 enzyme, a key regulator of the ubiquitylation pathway [1]. This results in impaired cellular protein debris removal process presenting with vacuolization in the myeloid and erythroid precursors.
Patients with this syndrome were characterized by recurrent fevers, neutrophilic dermatosis, leukocytoclastic vasculitis, polyarteritis nodosa, ear and nose chondritis, venous thromboembolism, pulmonary infiltrates, macrocytic anemia, elevated acute-phase reactants, MDS, multiple myeloma, and bone marrow vacuolization restricted to myeloid and erythroid precursor cells [1]. Lack of response to disease modifying anti-rheumatic drugs (DMARDs), biologic agents and target synthetic DMARDs has been observed in these patients. Only high-dose glucocorticoids ameliorated severe inflammatory symptoms [1]. Bone marrow transplantation and gene-editing have been suggested as therapeutic options [1].
At 8 months of follow-up, our patient is currently clinically stable with gradual improvement in his symptoms without intervention. Hemoglobin has been stable in the range of 10.0 - 11.0 g/dL. He has not required red blood cell transfusion support for his anemia and he does not currently have other cytopenias. Systemic therapy for MDS is not currently warranted. Leukocytoclastic vasculitis rash has resolved without any oral medications and topical steroid cream resulted in symptomatic relief. He continues to have fatigue but fevers and body aches are improved. ESR and CRP have also improved (60 mm/h and 2.4 mg/dL, respectively) though they remain elevated. Pulmonary inflammation has been reported in 76% VEXAS patients [1]. However, due to the asymptomatic nature of the pulmonary nodules and absence of antineutrophil cytoplasmic antibodies, a lung biopsy is not recommended at this time.
Based on the study by Ferrada et al (2021), VEXAS patients who become transfusion-dependent and have a valine amino acid substitution at codon 41 have the highest risk of death (50%) compared to patients with leucine (18%) or threonine (22%) substitution [2]. Median survival from symptom onset in this study was 10 years. Our patient had leucine substitution of methionine at codon 41 which may explain his relatively indolent course thus far, and he may have a better prognosis.
In summary, patients with auto-inflammatory rheumatologic conditions associated with myelodysplasia and notable vacuoles in the myeloid and/or erythroid precursors should have VEXAS considered in the differential diagnosis and screened for UBA1 mutation. Because of the associated risk of bone marrow failure and hematological malignancies, it is prudent to continue close monitoring for disease progression in these patients. Some patients may have a less aggressive course of disease than initial case studies suggested, and specific UBA1 mutation variants may be prognostic. Certainly, additional data and long-term follow-ups are needed to further validate these findings.
Acknowledgments
We thank Dr. Michael Hew (Department of Pathology, Medical City Lewisville Hospital in Lewisville, TX).
Financial Disclosure
The authors have no financial disclosure to report.
Conflict of Interest
The authors have no conflict of interest to disclose.
Informed Consent
The authors have obtained informed consent from the patient.
Author Contributions
VT and MP both wrote and reviewed the final paper.
Data Availability
The authors declare that data supporting the findings in this report are available in this article.
| References | ▴Top |
- Beck DB, Ferrada MA, Sikora KA, Ombrello AK, Collins JC, Pei W, Balanda N, et al. Somatic Mutations in UBA1 and Severe Adult-Onset Autoinflammatory Disease. N Engl J Med. 2020;383(27):2628-2638.
doi pubmed - Ferrada M, Savic S, Alessi H, Ospina D, Wilson L, Goodspeed W, et al. Genotype and transfusion dependence predicts mortality in VEXAS syndrome, a newly described disease with overlap inflammatory and hematologic features [abstract]. Arthritis Rheumatol. 2021;73(suppl 10):1426.
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.