

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://www.thejh.org |
Review
Volume 11, Number 6, December 2022, pages 197-209

Acute Myeloid Leukemia Following Myeloproliferative Neoplasms: A Review of What We Know, What We Do Not Know, and Emerging Treatment Strategies
Zoe McKinnella ![]() , Daniel Karela, Daniel Tuerffa, Marwa SH Abrahima, Samah Nassereddinea, b
, Daniel Karela, Daniel Tuerffa, Marwa SH Abrahima, Samah Nassereddinea, b
aDepartment of Hematology and Oncology, George Washington University Hospital, Washington, DC, USA
bCorresponding Author: Samah Nassereddine, Department of Hematology and Oncology, George Washington University and George Washington Cancer Center, Washington, DC, USA
Manuscript submitted September 2, 2022, accepted October 15, 2022, published online December 1, 2022
Short title: A Review of Post-MPN AML
doi: https://doi.org/10.14740/jh1042
- Abstract
- Introduction
- Clinical Risk Factors for LT
- Molecular Risk Factors for LT
- Clonal Evolution of MPNs to AML: There Is Not a Clear Path to LT
- Risk Stratification and Response Criteria for Post-MPN AML
- Little Therapeutic Progress Has Been Made
- Standard Treatment Options: Medically Fit Patients
- Standard Treatment Options: Medically Unfit Patients
- Targeted Treatment Options With Negative Results to Date
- Novel Treatment Options That Have Shown Early Promise
- Preclinical Studies
- Conclusions
- References
| Abstract | ▴Top |
Acute myeloid leukemia (AML) arising from myeloproliferative neoplasms (MPNs) represents a small subtype of secondary AML (sAML). This entity is well known to be associated with poor responses to available treatment options and dismal outcomes. To date, there are no standardized treatment options and there has been very little therapeutic advancement in recent years. This is a stark contrast to other subsets of AML for which there have been significant advances in therapeutic approaches, especially for patients with targetable mutations. We aim to focus our review on the incidence, risk factors for leukemogenesis, pathogenesis, molecular landscape, and emerging therapeutic options in post-myeloproliferative neoplasm acute myeloid leukemia (post-MPN AML).
Keywords: Myeloproliferative neoplasms; Acute myeloid leukemia; Myelofibrosis; Allogeneic stem cell transplantation; Chemotherapy; Hypomethylating agents
| Introduction | ▴Top |
Philadelphia chromosome negative myeloproliferative neoplasms are chronic myeloid disorders which exhibit terminal myeloid expansion in the peripheral blood and are most commonly associated with mutations in the JAK/STAT pathway [1]. They include polycythemia vera (PV), essential thrombocytosis (ET), primary myelofibrosis (PMF) and pre-fibrotic myelofibrosis [2, 3]. They are characterized as chronic diseases and, as such, portend an increased risk for both thrombosis and bleeding, as well as risk of leukemic transformation (LT) [4, 5]. It is known that both genomic and clinical features play a substantial role in progression. Further genomic characterization of these patients can help to risk stratify and predict which patients are more likely to transform to a leukemic phase [6].
In the leukemic phase, the molecular and morphologic features of this subset of patients are very different from primary acute myeloid leukemia (AML) and other subsets of secondary AML [7]. Patients often harbor p53 mutations and high-risk cytogenetics and lack mutations which augur favorable risk such as NPM1 and FLT3 [8].
There are no treatments which significantly prolong survival apart from allogeneic stem cell transplantation (allo-SCT) [9]. In a retrospective analysis of 91 patients with AML arising from myelofibrosis, the median survival from the time of LT was 2.6 months [10], and the outcomes for those who are able to proceed to allo-SCT are inferior to patients with de novo AML [11]. Further progress is needed to identify which patients are most likely to progress from MPNs to acute leukemia, how to prevent such progression, and how to treat the patients who do progress more effectively [11].
| Clinical Risk Factors for LT | ▴Top |
Factors that have been implicated in the risk of LT include MPN subtype, exposure to prior therapies, clinical and molecular factors. In general, risk for LT varies by MPN subtype. PMF has the highest risk of LT (5.8-20.6% risk in 10 years) followed by PV (2.3-8.7% risk in 10 years). ET has the lowest risk of transformation (0.7-4% risk in 10 years) [12-16].
Historically, there was concern amongst physicians about the impact of precedent therapies on LT in chronic phase MPN. More recently, studies have evaluated whether any association exists. Therapies evaluated included radioactive phosphorous P32 (at a dose of more than 100 MBq), alkylators (mainly busulfan and pipobroman), erythroid stimulating agents (ESAs), and hydroxyurea. MPN patients treated with radioactive phosphorous and alkylators were found to have a statistically significant increased risk for LT [17, 18]. Studies have been inconsistent as to whether or not ESAs increase the risk of LT in MPN patients and the use ESAs is not discouraged in MPN patients [19-21]. Hydroxyurea has consistently shown to have no increased risk of LT and remains a standard treatment for MPN patients [17, 18, 20].
Many clinical factors have been associated with an increased risk for LT in MPN patients including advanced age, cytopenias, and transfusion dependence [16]. More specifically, for PMF patients, thrombocytopenia (with a platelet count < 100 × 109/L) and peripheral blood blasts of 3% or more at diagnosis were found to be significant and independent predictors of LT [20]. In addition, time dependent factors which were prognostic for LT in MF patients include time to onset of all of the following: anemia (hemoglobin < 10 g/dL), leukocytosis (white blood cell (WBC) > 30 × 109/L), thrombocytopenia (platelet count < 150 × 109/L) and chemotherapy initiation [22]. For ET, a hemoglobin level below normal (< 12 g/dL in females and < 13.5 g/dL in males) was identified as an independent risk factor for both decreased survival and LT [23]. For PV, older age (> 70 years) was identified as the most significant risk factor for LT with a hazard ratio (HR) of 4.30 [18].
While these clinical risk factors are significant, molecular risk factors play a more significant role [16].
| Molecular Risk Factors for LT | ▴Top |
Improvements in cytogenetic and next generation sequencing (NGS) testing over the past several decades have led to advancements in our abilities to predict which MPN patients are at highest risk for LT [24].
The Dynamic International Prognostic Scoring System (DIPSS) was initially published in 2010 as a prognostic scoring system for PMF based on data from 1,054 patients diagnosed in the years 1980 - 2007. This scoring system identified clinical factors including age, constitutional symptoms, WBC, hemoglobin, and blasts as prognostic parameters. It categorized patients into four risk groups ranging from high-risk (median overall survival of 26 months) to low-risk (median overall survival of 135 months). Cytogenetic abnormalities were not incorporated into this model secondary to limited sampling at that time [25]. This scoring system was later refined to include karyotype, platelet count, and transfusion status. This model also found that thrombocytopenia and unfavorable karyotype predicted leukemia free survival (10-year risk of 31% vs. 12%; HR: 3.3; 95% confidence interval (CI): 1.9 - 5.6) [26].
Since the development of the DIPSS, further work has been done to determine the risk of LT in MPN patients. Tefferi et al [27] developed a prognostic model to predict death and LT within 2 years of diagnosis for PMF patients. This model found that risk factors associated with 2-year mortality of at least 80% included monosomal karyotype, inv(3)/i(17q) abnormalities, or any two of the following: circulating blasts > 9%, leukocytes ≥ 40 × 109/L, or other unfavorable karyotype. Additionally, it showed that 26% of patients with a monosomal karyotype and 25% of patients with inv(3)/i(17q) abnormalities underwent LT. This study led to the development of a new very high-risk category [28]. This cytogenetic risk stratification for PMF patients was again revised to a three-tiered risk model which not only predicted overall survival (OS) but was also effective in predicting risk to LT. This model defined very high risk as abnormalities of -7, i(17q), inv(3)/3q21, 12p-/12p11.2, 11q-/11q23 or autosomal trisomies excluding + 8/+ 9 (e.g., +21, +19. Patients in the very high-risk category had a 4.4 increased risk of LT [27].
Independent of these prognostic models, several mutations in activating signaling pathways and epigenetic and transcriptional regulators have been implicated in the LT of post-myeloproliferative neoplasm acute myeloid leukemia (post-MPN AML) notably ASXL1/SRSF2 and TP53 [27, 29]. An analysis of 73 patients with post-MPN AML identified TP53, SRSF2 and TET2 as independent negative prognostic factors when controlling for preexisting MPN and treatments received. This analysis found that the time to AML transformation from MPN diagnosis was 51.2 months in patients with SRSF2 mutations as compared to 133.8 months in SRSF2 unmutated patients [30].
Table 1 shows the mutations that are implicated in post-MPN AML and have a frequency of 15% of higher [30-35].
 Click to view | Table 1. Mutations That Are Implicated in Post-MPN AML and Have a Frequency of 15% of Higher |
Grinfeld et al sequenced coding exons from 2,035 patients with MPNs and identified eight distinct clinical phenotypes. They found that TP53 and MPL were associated with higher risk of leukemia transformation with a HR 15 and 8.6, respectively [31]. Their work led to the development of a personalized MPN risk calculator (“New Classification for Myeloproliferative Neoplasms,” 2018b, https://www.sanger.ac.uk/science/tools/progmod/progmod/). Further validation of this model, and others, could have significant clinical implications by guiding MPN treatment to either focus on symptoms or to focus on more intensive treatments to prevent LT.
| Clonal Evolution of MPNs to AML: There Is Not a Clear Path to LT | ▴Top |
The pathogenesis of myeloproliferative disorders is well established. A single point activating mutation in the Janus protein tyrosine gene JAK2 (JAK2 617F) is the main driver of MPNs. The JAK/STAT signaling pathway is essential in the normal hematopoiesis and the regulation of the micro-environment of the bone marrow by regulating cytokine release. The occurrence of mutations that activate this pathway lead to a cascade of proliferation and dysregulation of the hematopoiesis by creating a pro-inflammatory state. This pro-inflammatory state can lead to leukemogenesis.
In contrast, we do not have a clear understanding of the pathogenesis of LT. It is most likely a dynamic process involving the acquisition of mutations and epigenetic alterations which result in clonal selection. In 50% of cases, MPN patients lose the JAK2-V617F mutation during this process [36].
It is known that high molecular risk (HMR) mutations, such as ASXL1, EZH2, SRSF2 and IDH1/2 or TP53 have been associated with adverse prognosis; however, the order in which these mutations occur has yet to be determined [37].
Lundberg et al used NGS to follow somatic mutations in serial samples from patients with MPN. They focused on patients carrying mutations in epigenetic modifier genes for their clonal analysis. They found that mutations in TET2 and DNMT3A were acquired before JAK2-V617F and mutations in IDH1 occurred after JAK2-V617F. Mutations with low allelic burden frequency including TP53 and KRAS/NRAS were considered late events in MPN pathogenesis. They also observed that TP53 mutations were often present in a heterozygous state in chronic phase MPN. However, once the wild type allele was lost, the TP53 expanded rapidly, often leading to LT [38].
Several studies, both in vivo and in vitro, have shown that somatic mutations in TP53 lead to clonal dominance of JAK2-V617F/TP53-mutant leukemic cells and p53 loss is sufficient for inducing LT in JAK2-V617F-positive MPN [39, 40]. Courtier et al sequenced samples from 38 post-MPN AML patients. Their results suggested that mutations in TP53, DNA methylation and transcription factors were simultaneous with an evolution to AML. In contrast, mutations in RNA splicing, chromatin modifications, and signaling pathways were often found in chronic phase MPN samples, prior to LT and may increase risk [41].
Taken together, these findings suggest that serial molecular monitoring with NGS, particularly to detect TP53 mutations, may have a role in chronic phase MPN to assess disease evolution and potentially impact treatment.
| Risk Stratification and Response Criteria for Post-MPN AML | ▴Top |
It is difficult to apply conventional clinical factors used to risk stratify patients with AML (age, karyotype, ELN2017 prognostic classification, treatments received, treatments response, transplant status) to post-MPN AML patients, because their disease differs biologically from de novo disease. And due to the high frequency of older age, complex karyotypes and TP53 mutations, the vast majority of patients would be categorized as having adverse risk [42]. While it is true that post-MPN AML carries a dismal prognosis, there may still be heterogeneity amongst these patients, and we currently do not have a standardized way to risk stratify them separately from de novo AML.
Furthermore, only recently, was a standardized response criterium established for post-MPN AML. This updated criterium reflect improvement in both the leukemic and MPN component of disease. For example, a complete molecular response is defined as a complete remission (CR) of both leukemia and MPN without detectable molecular markers associated with either leukemia or MPN. It incorporates lab values, bone marrow biopsy results, spleen size, cytogenetics and molecular markers [43]. This set of response criteria may have to be updated in the future as we learn more about the pathogenesis of post-AML, but in the meantime, it allows for an even playing field for new therapeutics.
| Little Therapeutic Progress Has Been Made | ▴Top |
There has not been an improvement in OS for post-MPN AML in several decades. Tefferi et al demonstrated in a 2018 retrospective study that treatment-specified 3-year and 5-year survival rates were 32% and 10%, respectively for patients receiving allo-SCT, 19% and 13% respectively for chemotherapy-induced patients achieving CR/CRi (CR with incomplete cell count recovery) (but were not transplanted), and 3-year survival rate was only 1% in the absence of both allo-SCT and induction [44].
The reasons for these grim results are multifactorial. Patients with post-MPN AML tend to be older, with more comorbidities, and have a worse performance status at baseline. Second, they tend to have poor risk molecular features compared to de novo AML. Third, these molecular changes may confer chemotherapy resistance. One particular described mechanism of chemoresistance is the overexpression of multidrug resistant (MDR) genes resulting in a rapid drug efflux [45]. For instance, ASXL1, commonly detected in MPN clones, predicts resistance to chemotherapy and poorer outcomes [46].
Finally, post-MPN patients have been traditionally excluded from many AML treatment trials. Currently available data for post-MPN AML come from studies that are mostly retrospective and have had widely varying results, highlighting the need for clinical trials. A phase III study published in 2021 demonstrated that CPX-351 (liposomal daunorubicin and cytarabine) had superior 2-year OS in patients with secondary or high-risk cytogenetic AML when compared with standard 3 + 7 induction therapy [47]. Post-MPN AML patients were excluded from this study.
Table 2 shows the comparison of median overall survival among different treatment regimens and studies [4, 9, 10, 44, 47-56].
Click to view | Table 2. A Comparison of Median Overall Survival Among Different Treatment Regimens and Studies |
| Standard Treatment Options: Medically Fit Patients | ▴Top |
Standard treatment regimens for medically fit patients involve induction chemotherapy and allo-SCT if available.
There are several chemotherapy regimens for induction chemotherapy suitable for post-MPN AML including daunorubicin + cytarabine (7 + 3) and mitoxantrone, etoposide, and cytarabine (NOVE-HIDAC) [7, 57].
A study published in 2013 comparing outcomes in treated patients with post-MPN AML showed that 77% of 39 patients treated with induction therapy were able to achieve response, defined as remission with count recovery, remission with incomplete count recovery or reversion to chronic MPN [49]. Of these patients induced, NOVE-HIDAC had superior rate of response, with 10/12 patients achieving response compared with 14/25 of those who received 7 + 3 therapy. Overall, those patients treated with induction therapy had improved 2-year survival (median overall survival 9.4 months vs. 2.3 months) compared with supportive therapy alone, although outcomes were worse compared with those who received stem cell transplantation. It has been well documented that induction therapy alone does not improve long-term survival.
Allo-SCT is currently the most effective treatment option for post-MPN AML and the only treatment that prolongs survival, regardless of the initial therapy utilized [53]. A retrospective study from 2012 showed that 6/8 patients who received transplantation achieved remission and were alive at 15 months. Notably, in this study 5/8 patients who underwent transplant had previously achieved CR or reversion to chronic phase MPN following induction chemotherapy, leaving three patients who had residual blasts in marrow or blood at the time of transplantation. Of these three patients with residual blasts, only one of the three remained alive in remission following transplant, indicating the importance of induction therapy [9]. Transplant remains a procedure associated with high morbidity and mortality limiting its utility in post-MPN AML patients especially, given their more advanced age, comorbidities, and tumor genetic complexity [44].
Furthermore, the role of transplant in improving OS is controversial. The post-MPN AML patients who are able to proceed with transplant are unique: they are fit enough to withstand the toxicities associated with allo-SCT and they have survived induction chemotherapy. In a retrospective analysis of post-MPN AML patients, using the Mantel-Byar test to control for immortality bias, allo-SCT failed to significantly improve OS [58]. There is also discussion about the timing of transplant and whether or not it should be considered in high-risk patients prior to LT. Interestingly, Marcault et al found that somatic mutations of nuclear factor erythroid 2 (NFE2) were detected at different time points during the MPN disease course and were associated with loss of treatment response [59]. Taken together, further work to identify signals to prompt transplant, such as NFE2 mutations, before MPN patients transform to leukemia may be a promising area of future research.
The highly toxic nature of this treatment and its associated morbidity and mortality makes it clear that new treatment modalities are desperately needed to improve survival and quality of life in patients with post-MPN AML.
| Standard Treatment Options: Medically Unfit Patients | ▴Top |
Post-MPN AML patients who are medically unfit to receive induction chemotherapy, due to suboptimal performance status or medical comorbidities, are treated similarly to medically unfit patients with de novo AML [52]. They are treated with low-intensity chemotherapy regimens made up of a hypomethylating agent (HMA), and if available, the BCL-2 inhibitor venetoclax [48]. The outcomes for these patients treated with hypomethylating agents is similar to those treated with induction chemotherapy. It is thought that they are effective because they dampen hypermethylation which is thought to be central to the pathogenesis of post-MPN AML. More specifically, DNA hypermethylation of the p15INK4B and p16INK4A genes located on chromosome 9p21 and retinoic acid receptor β have been reported to be crucial in the pathogenesis of accelerated-phase MPN (MPN-AP) and post-MPN AML [50].
Since these results are similar to giving standard induction chemotherapy, hypomethylating agents are considered a good option for all patients, and especially those who may not be transplant eligible. There are even several reports showing some improvement in response rates and survival benefit with the use of HMA [16, 44, 48]. Patients who respond to HMAs may have recurrence of their MPN phenotype such as thrombocytosis and polycythemia, with reoccurrence in 39% of patients in one study [54].
For patients considered frail, supportive therapy is often the treatment of choice (Fig. 1) [4, 10, 16, 48, 49, 53].
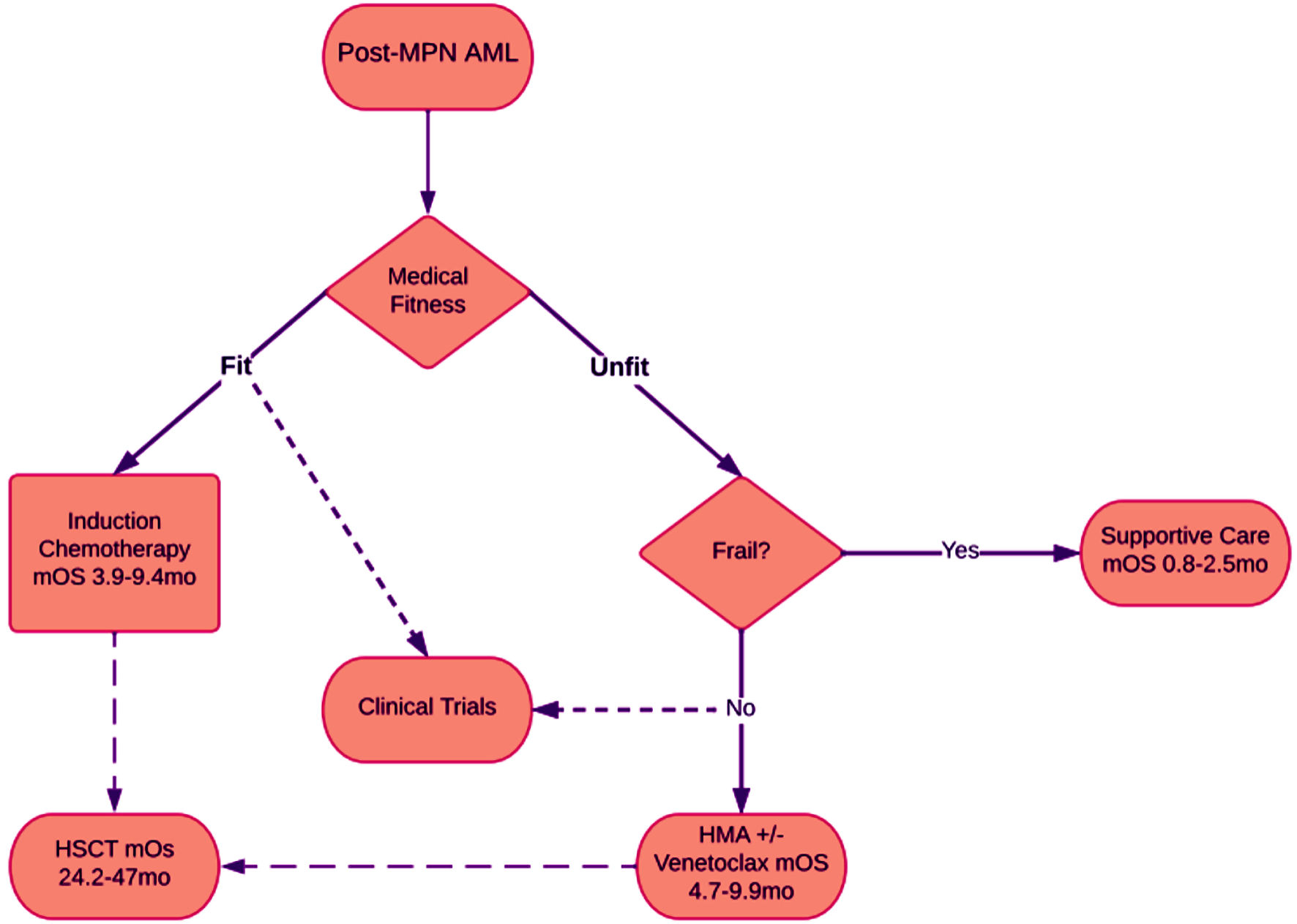 Click for large image | Figure 1. Flowchart showing treatment options for patients with post-MPN AML. Patients are assessed for medical fitness, which can be represented with Eastern Cooperative Oncology Group (ECOG) performance status. Generally, patients with ECOG 0 - 1 can be considered fit. Patients with ECOG 2 - 3 are often considered unfit. Patients with ECOG 4 are considered frail. Most often fit patients are considered for clinical trials, however “unfit” are sometimes also included, and frail patients are rarely included. Median overall survival was gathered from previous clinical trials with ranges displayed. mOS: median overall survival; mo: months. |
| Targeted Treatment Options With Negative Results to Date | ▴Top |
JAK2 inhibitors
JAK inhibitors were initially developed in 2005 prompted by the discovery of the JAK2-V617F driver mutation in MPNs. JAK2 inhibitors have shown clinical benefit by alleviated symptoms in MPN patients but have not been shown to meaningfully prolong survival in PMF [60, 61]. JAK2 inhibitors have been postulated to have a role in treating post-MPN AML and are still being studied, both alone and in combination with HMAs. So far, response rates are not promising.
Mechanistically, mutations in the JAK/STAT pathway leading to constitutively STAT activation are well characterized in leukemia. The constitutive activation of STAT5 in particular has been shown to play a key role in the malignant transformation to post-MPN AML [62]. However, the JAK2 clone is often lost in patients who develop post-MPN AML and is not thought to have a role in the transformation of acute leukemia [4].
Ruxolitinib was approved in 2011 for the treatment of intermediate and high-risk myelofibrosis [50]. A phase II study of ruxolitinib in refractory post-MPN AML showed that ruxolitinib as a single agent had a modest antileukemic effect with 3/18 patients demonstrating a significant response [63]. A phase II study of 25 patients with MPN-AP/post-MPN AML were treated at the recommended phase II dose of ruxolitinib 25 mg twice daily for the induction cycle followed by 10 mg twice daily for subsequent cycles in combination with decitabine 20 mg/m2 for 5 consecutive days in a 28-day cycle. The median overall survival was 9.5 months similar to treatment with an HMA alone [56].
Interestingly, ruxolitinib may have a role in alleviating symptoms and improving performance status in patients who are allogenic hematopoietic stem cell transplantation (allo-HSCT) transplant candidates. In a case report of six patients who got ruxolitinib in combination with intensive chemotherapy, three patients underwent allo-HSCT [64]. There is also an ongoing clinical trial evaluating the role of decitabine in combination with a JAK-inhibitor as a bridge to allo-HSCT (NCT04282187).
Fedratinib is a selective oral JAK2 inhibitor approved for the treatment of adults with intermediate or high-risk myelofibrosis. In preclinical models, fedratinib blocked phosphorylation of the STAT5 pathway and was found to exert off target inhibitory activity against bromodomain-containing protein 4 (BRD4), which, in turn, was found to attenuate disease burden and reverse bone marrow fibrosis [65]. There is an ongoing phase II study (NCT04282187) evaluating the use of fedratinib with decitabine before transplant in patients with accelerated/blast phase MPN.
BH3-mimetics/BCL-2 inhibitors
Venetoclax (ABT-199), a BH3-mimetic and highly selective protein B-cell lymphoma 2 (BCL-2) inhibitor, was approved in November 2018 by the US Food and Drug Administration (FDA) for the treatment of AML in adult patients aged 75 years or older, or unable to tolerate intensive induction chemotherapy, in combination with either hypomethylating agents or low-dose cytarabine [66]. While the use of venetoclax has shown benefit in de novo AML and secondary AML (MRC or therapy related), its effect is unclear in patients with post-MPN AML.
It is suggested that cells chronically treated with JAK2i reversibly hyperactivated the JAK2/STAT5 signaling axis, with increased expression of Bcl-xL, and surprisingly, decreased levels of Bcl-2. This can confer resistance to venetoclax. Results from retrospective studies have been disappointing and not only reflected no difference in outcomes but also increased toxicities from adding venetoclax to HMA. A retrospective case series comparing venetoclax (Ven) plus HMA to historic controls treated with HMA alone demonstrated that the CR/CRi rate and median survival (8 vs. 5.5 months) were more favorable with Ven + HMA, but without significant difference in OS [52]. A different single-center retrospective study of 31 patients (14 newly diagnosed and 17 relapsed/refractory (R/R)), found that venetoclax did not improve outcomes and was associated with significant morbidity secondary to adverse effects. Specifically, they found no improvement in clinical remission rates when venetoclax was added to a single agent HMA treatment in the front-line setting, and none of the 10 patients treated with a combination of HMA and venetoclax exhibited any clinical response. Furthermore, adding venetoclax was associated with significant myelosuppression and mortality. Grade 3 or high infections were observed in 86% of the patients treated in the frontline setting and 93% of the patients getting treated for relapsed or refractory disease [67].
Navitoclax (ABT-737), an orally available BCL-XL/BCL-2 inhibitor appears to be a promising targeted agent in MPN clones [68] and has demonstrated the ability to overcome JAK inhibitor resistance in preclinical models. Navitoclax binds with high affinity to BCL-2 family proteins including BCL-X, BCL-2 and BCL-2 disrupting interactions with proapoptotic factors and promotes apoptosis of malignant cells. Preclinical data have indicated that ruxolitinib and navitoclax act synergistically. Ruxolinitib suppresses BCL-X and MCL-1 expression and then allows navitoclax to inhibit the remaining BCL-X and BCL-2 [69].
A phase II study found that adding navitoclax to JAK2 inhibitors has disease modifying activity in patients with PMF with progression or refractory to JAK inhibition [70], suggesting that this treatment would be effective in patients with post-MPN AML. Of note, prolonged thrombocytopenia has limited the use of navitoclax [71]. In this study, thrombocytopenia was noted in 88% of patients. However, it was described as manageable, reversible on dose reduction/interruption of navitoclax or ruxolitinib and it was not associated with major bleeding events.
| Novel Treatment Options That Have Shown Early Promise | ▴Top |
IDH1/2 inhibitors
Mutations in isocitrate dehydrogenase genes (IDH1 and IDH2) lead to abnormal epigenetic regulation in AML cells and occur in up to 30% of de novo AML cases [72]. Interesting, the occurrence of IDH1/2 mutations increase from about 1% to 4% in chronic-phase MPN (MPN-CP) to about 22% in post-MPN AML, strongly evidencing their role in MPN progression [73]. Enasidenib and ivosidenib are small molecule inhibitors of mutant IDH2 and IDH1, respectively. They have been approved by the FDA for relapsed or refractory AML associated with an IDH mutations, as well as first-line treatment for elderly (≥ 75 years) or unfit patients for ivosidenib. Results of a retrospective study of seven patients with IDH1 mutations treated with IDH1/2 inhibitor-based regimens in the frontline setting are promising. Of these patients, three patients achieved a clinical response with undetectable IDH1/2 mutation by NGS and one patient achieved measurable residual disease negativity by flow cytometry [74]. There is a need for further clinical studies addressing the role of IDH inhibitors in post-MPN AML patients.
PARP inhibitors
Poly-(ADP) ribose polymerase (PARP) represents a family of enzymes critical for sensing DNA damage and repairing both single and double stranded DNA breaks, and thus may acts as sensitizer to chemotherapy [75]. Olaparib, rucaparib, niraparib and talazoparib are currently FDA approved for various solid tumors. It has previously been shown that monotherapy with PARP-1 inhibition has benefit in patients with BRCA1/2 associated solid epithelial malignancy such as recurrent ovarian cancer and it has been hypothesized that they may also have a role in AML in those with certain cytogenetic characteristics [76, 77].
Preclinical models suggest certain mutations, common in post-MPN AML, pre-dispose sensitivity to PARP inhibition, for example RUNX1 mutations [74]. MPN samples showed increased sensitivity to the PARP inhibitors veliparib and olaparib compared with normal myeloid progenitors, highlighting the potential for this class of medication for use in post-MPN AML [78]. Furthermore, PARP-1 inhibition combined with HMA has been shown to improve treatment of myelodysplastic syndromes (MDS) and AML mediated by preventing efficient base excision repair through disruption of the relocation of XRCC1 to DNA damage sites [79]. Because this mechanism was studied in de novo AML, it remains to be determined if there is any benefit in post-MPN AML.
PARP inhibitors may have a role in pre-transplant conditioning. A preclinical study described a novel pre-transplantation conditioning regimens with combinations of PARP-1 inhibition and reduced doses of alkylators, such as busulfan and melphalan, for high-risk MPNs or post-MPN AML. This study demonstrates that combination of melphalan and veliparib improved survival in a JAK2-V617f post-MPN AML xenotransplant mouse model [80]. This study also demonstrated that these same two medications exerted a synergistic anti-proliferation effect of CD34+ cells derived from patients with PMF. This concept is further corroborated by a study in 2018 combining the JAK-1/2 inhibitor ruxolitinib and PARP-1 inhibition resulting in lethal DNA damage to MPN cells bearing the JAK2, CALR or MPL mutation [81].
BET inhibitors
The family of bromodomain and extra terminal (BET) proteins (including BRD2, BRD3, BRD4 and BRDT) are “chromatin reader” proteins that contain N-terminal, double-tandem bromodomains that bind to the acetylated lysine on the nucleosomal histones. They are associated with multiple proliferative cell signalizing pathways, such as NF-κB [82]. There are 97 target pathways found to be affected by BET inhibition, although the most common were BRD4 and MYC [83]. A 1b dose escalation trial recently published shows BET inhibition (RO6870810) in combination with venetoclax and rituximab and demonstrated both safety and efficacy in diffuse large B-cell lymphoma (DLBCL) [84], and another preclinical study showed the combination of BRD4 antagonist and histone deacetylase inhibitor are active against human AML cells [81, 85]. This pathway has been studied as a therapeutic target in post-MPN AML [86].
Inhibitors of BET (BETis) show promising apoptotic activity in MPN blast derived cells in preclinical data [82]. BET inhibitor JQ1 exhibited lethal synergistic effect when combined with JAK inhibitors in patient derived post-MPN AML progenitor cells and attenuated the protein expressions of proliferative signals (such as c-MYC, p-STAT5, Bcl-xL, CDK4/6, PIM1 and IL-7R) [87]. It concomitantly induced the levels of anti-proliferative signaling (including HEXIM1, p21 and BIM in MPN) in an erythroleukemia cell line.
Based on promising preclinical data, there are several ongoing clinic trials evaluating the role of BET inhibition in MPNs. These studies mainly evaluate the BET inhibitor pelabresib (CP-0610) in combination with JAK inhibitors (NCT04603495, NCT02158858).
| Preclinical Studies | ▴Top |
Mutations in chromatin modifiers and transcription factors downstream of JAK2, MPL and CALR contribute to a dysregulated transcriptome and to the aggressive phenotype and therapy refractoriness in post-MPN AML cells. Many novel treatments which have shown early promise aim to target these downstream transcription factors and epigenetic regulators.
TP53
TP53 is a crucial tumor suppressor gene widely known as “the guardian of the genome”, stimulating cell cycle arrest, DNA repair and apoptosis in situations of cellular stress. TP53 is mutated in approximately 20% of post-MPN AML cases and is much more common than in de novo AML [88]. TP53 expression is also commonly inactivated by the overexpression of its inhibitors MDMX (which include MDM2 and MDM4) [89]. The mechanisms of MDM2 and MDM4 complement each other. MDM4 mainly regulates p53 activity and MDM2 regulates p53 stabilization [89]. Like TP53 mutations, the overexpression of MDMX also confers a worse prognosis [32]. Preclinical studies suggest that MDM4 plays a critical role in MPN disease progression and post-MPN AML providing evidence of the potential for the usefulness of targeting the TP53 pathway [88]. Novel therapies targeting the TP53 pathway have been generally focused on the inhibition of MDMX and/or inhibiting its interaction with p53.
A preclinical study demonstrated that MDM2 inhibition combined with BET (BRD4) inhibition demonstrated an enhanced cytotoxic effect in AML cell lines, murine model and primary human blast cells [90]. Another preclinical study by Wang et el in 2021 shows an exciting proof of concept for combination of MDM2 inhibitors and check point inhibitors such as PD-1, as MDM2 inhibition was shown to alter the immune tumor microenvironment favorably (i.e., increases the CD8/Treg ratio, upregulates dendritic cells) [91].
RG7112 (Hoffmann La Roche RO5045337) is the first studied MDM2 inhibitor in human clinical trials and it demonstrated antitumor activity through reactivation of P53 and increased expression of downstream pro apoptotic proteins [66]. When studied in AML, RG7112 as monotherapy and in combination with low-dose cytarabine showed some response but resulted in prolonged dose-dependent toxicity [92]. A phase I clinical trial across multiple malignant diseases investigated the recommended dose for expansion for siremadlin, a p53-MDM2 inhibitor. This study included 93 patients with hematologic malignancies and found increased response rates, although also increased toxicity when compared to solid tumors, the most common being myelosuppression [93].
More recently, the phase III MiRROS trial of 436 patients evaluated the efficacy and safety of the small-molecule MDM2 antagonist idasanutlin plus cytarabine in patients with R/R AML. The trial did show an improved overall response rate 38.8% vs. 22.0% in the treatment arm. However, despite improved overall response, adding idasanutlin to cytarabine did not improve OS or clinical remission rates [94].
Despite the early clinical promise of p53 activation via MDMX pathway inhibition, there is concern that this new therapeutic strategy may stimulate the expansion of TP53 mutated subclones. While TP53 mutations are rarely detected in chronic phase MPN, they are found at very low frequency (VAF) in 14.7% of chronic phase patients, and the presence of minor TP53 mutations increases with age [95]. Marcellino et al found that idasanutlin (an MDM2 antagonist) is associated with expansion with the TP53 subclone. They found that after getting idasanutlin, five patients had accumulated 12 TP53 subclones and only one patient was noted to have a TP53 mutation prior to getting treatment [96]. These findings should be kept on the horizon and studied on a larger scale to determine whether or not this is clinically significant and whether patients acquire de novo TP53 mutations with idasanutlin treatment.
There are also early phase trials ongoing evaluating APR-246 (eprenetapopt), a small molecule that restores wild-type p53 functions in TP53-mutant cells. A phase Ib/II trial of 55 patients (11 with AML) of APR264 (eprenetapopt) given in combination azacitidine in patients with TP53 mutations had promising results with an overall response rate and CR rate for patients with AML of 64% (n = 7) and 36% (n = 4), respectively [97]. There is an ongoing phase III trial (NCT03745716) of azacitidine +/- aza + APR-246 for MDS patients. It would be interesting to replicate this study in patients with TP53 mutated post-MPN AML.
CDK9 inhibition
CDK9 is a cyclin dependent kinase that controls transcriptional processes [98]. Significantly enhanced CDK9 activity has been observed in many cancer types and inhibition of CDK9 is an evolving area of interest. CDK9 activity helps promotes growth and survival of AML cells by maintaining production of proteins such as MCL-1 and c-myc [2]. Preclinical studies have demonstrated that targeted inhibition of CDK9 exerts lethal in vitro activity against post-MPN AML cells. Early phase clinical trials of CDK9i as a monotherapy (NCT03263637) are underway.
| Conclusions | ▴Top |
Post-MPN AML is a distinct entity of AML with poor clinical outcomes and limited treatment options. While we know that a single point activating mutation in the JAK2 617F is the main driver of MPNs, we do not have a clear understanding of the pathogenesis of post-MPN AML. Mutations in activating signaling pathways, epigenetic and transcriptional regulators have been found to be associated with post-MPN AML such as TET2, TP53 and SRSF2; and new work is being done to help identify which patients are at highest risk for transformation from MPN to AML based primarily on their genetic profile. Traditional treatment options for acute leukemia such as induction chemotherapy and HMA with venetoclax have not been shown to significantly prolong OS, and allo-SCT remains the only effective treatment. However, early studies targeting pathways that lead to epigenetic dysregulation such as IDH1/2 and BET inhibitors have shown some promise. In sum, despite some recent progress, more work is needed to further understand the pathogenesis of post-MPN AML in hopes that this would lead to improved treatment options.
Acknowledgments
None to declare.
Financial Disclosure
The authors declare that they do not have a financial relationship with any commercial entity that has an interest in the subject of this manuscript.
Conflict of Interest
The authors declare that they do not have a conflict of interest.
Author Contributions
All authors participated in this review. The authors confirm contribution to the paper as follows. Topic and outline: Samah Nassereddine. Research collection, analysis, and interpretation: Zoe McKinnell, Daniel Karel, Daniel Tuerff, Marwa SH Abrahim and Samah Nassereddine. Draft manuscript preparation: Zoe McKinnell and Daniel Karel. Figure and table preparation: Daniel Tuerff. All authors reviewed the results and approved the final version of the manuscript.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
Abbreviations
AML: acute myeloid leukemia; MPN: myeloproliferative neoplasms; sAML: secondary AML; post-MPN AML: post-myeloproliferative neoplasm acute myeloid leukemia; PV: polycythemia vera; ET: essential thrombocytosis; PMF: primary myelofibrosis; alloSCT: allogeneic stem cell transplantation; LT: leukemic transformation; JAK2 617F: Janus protein tyrosine gene JAK2; CR: clinical remission; MDR: multidrug resistant; HMA: hypomethylating agent; MPN-AP: accelerated-phase MPN; ECOG: Eastern Cooperative Oncology Group; BRD4: bromodomain-containing protein 4; BCL-2: B-cell lymphoma 2; MPN-CP: chronic-phase MPN; PARP: poly-(ADP) ribose polymerase; BET: bromodomain and extra terminal; NGS: Next Generation Sequencing; ESA: erythroid stimulating agent
| References | ▴Top |
- Fowlkes S, Murray C, Fulford A, De Gelder T, Siddiq N. Myeloproliferative neoplasms (MPNs) - Part 1: An overview of the diagnosis and treatment of the "classical" MPNs. Can Oncol Nurs J. 2018;28(4):262-268.
doi pubmed - Anshabo AT, Milne R, Wang S, Albrecht H. CDK9: a comprehensive review of its biology, and its role as a potential target for anti-cancer agents. Front Oncol. 2021;11:678559.
doi pubmed - New Classification for Myeloproliferative Neoplasms. Cancer Discov. 2018;8(12):OF1.
doi pubmed - Passamonti F, Rumi E, Pietra D, Elena C, Boveri E, Arcaini L, Roncoroni E, et al. A prospective study of 338 patients with polycythemia vera: the impact of JAK2 (V617F) allele burden and leukocytosis on fibrotic or leukemic disease transformation and vascular complications. Leukemia. 2010;24(9):1574-1579.
doi pubmed - Iurlo A, Cattaneo D, Gianelli U. Blast transformation in myeloproliferative neoplasms: risk factors, biological findings, and targeted therapeutic options. Int J Mol Sci. 2019;20(8):1839.
doi pubmed - Klampfl T, Harutyunyan A, Berg T, Gisslinger B, Schalling M, Bagienski K, Olcaydu D, et al. Genome integrity of myeloproliferative neoplasms in chronic phase and during disease progression. Blood. 2011;118(1):167-176.
doi pubmed - Heaney ML, Soriano G. Acute myeloid leukemia following a myeloproliferative neoplasm: clinical characteristics, genetic features and effects of therapy. Curr Hematol Malig Rep. 2013;8(2):116-122.
doi pubmed - Marcellino BK, Hoffman R, Tripodi J, Lu M, Kosiorek H, Mascarenhas J, Rampal RK, et al. Advanced forms of MPNs are accompanied by chromosomal abnormalities that lead to dysregulation of TP53. Blood Adv. 2018;2(24):3581-3589.
doi pubmed - Cherington C, Slack JL, Leis J, Adams RH, Reeder CB, Mikhael JR, Camoriano J, et al. Allogeneic stem cell transplantation for myeloproliferative neoplasm in blast phase. Leuk Res. 2012;36(9):1147-1151.
doi pubmed - Mesa RA, Li CY, Ketterling RP, Schroeder GS, Knudson RA, Tefferi A. Leukemic transformation in myelofibrosis with myeloid metaplasia: a single-institution experience with 91 cases. Blood. 2005;105(3):973-977.
doi pubmed - Schmaelter AK, Labopin M, Socie G, Itala-Remes M, Blaise D, Yakoub-Agha I, Forcade E, et al. Inferior outcome of allogeneic stem cell transplantation for secondary acute myeloid leukemia in first complete remission as compared to de novo acute myeloid leukemia. Blood Cancer J. 2020;10(3):26.
doi pubmed - Cervantes F, Tassies D, Salgado C, Rovira M, Pereira A, Rozman C. Acute transformation in nonleukemic chronic myeloproliferative disorders: actuarial probability and main characteristics in a series of 218 patients. Acta Haematol. 1991;85(3):124-127.
doi pubmed - Barbui T, Thiele J, Passamonti F, Rumi E, Boveri E, Ruggeri M, Rodeghiero F, et al. Survival and disease progression in essential thrombocythemia are significantly influenced by accurate morphologic diagnosis: an international study. J Clin Oncol. 2011;29(23):3179-3184.
doi pubmed - Tefferi A, Rumi E, Finazzi G, Gisslinger H, Vannucchi AM, Rodeghiero F, Randi ML, et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia. 2013;27(9):1874-1881.
doi pubmed - Passamonti F, Rumi E, Arcaini L, Boveri E, Elena C, Pietra D, Boggi S, et al. Prognostic factors for thrombosis, myelofibrosis, and leukemia in essential thrombocythemia: a study of 605 patients. Haematologica. 2008;93(11):1645-1651.
doi pubmed - Tefferi A, Lasho TL, Guglielmelli P, Finke CM, Rotunno G, Elala Y, Pacilli A, et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016;1(1):21-30.
doi pubmed - Bjorkholm M, Derolf AR, Hultcrantz M, Kristinsson SY, Ekstrand C, Goldin LR, Andreasson B, et al. Treatment-related risk factors for transformation to acute myeloid leukemia and myelodysplastic syndromes in myeloproliferative neoplasms. J Clin Oncol. 2011;29(17):2410-2415.
doi pubmed - Finazzi G, Caruso V, Marchioli R, Capnist G, Chisesi T, Finelli C, Gugliotta L, et al. Acute leukemia in polycythemia vera: an analysis of 1638 patients enrolled in a prospective observational study. Blood. 2005;105(7):2664-2670.
doi pubmed - Rampal R, Mascarenhas J. Pathogenesis and management of acute myeloid leukemia that has evolved from a myeloproliferative neoplasm. Curr Opin Hematol. 2014;21(2):65-71.
doi pubmed - Huang J, Li CY, Mesa RA, Wu W, Hanson CA, Pardanani A, Tefferi A. Risk factors for leukemic transformation in patients with primary myelofibrosis. Cancer. 2008;112(12):2726-2732.
doi pubmed - Tam CS, Kantarjian H, Cortes J, Lynn A, Pierce S, Zhou L, Keating MJ, et al. Dynamic model for predicting death within 12 months in patients with primary or post-polycythemia vera/essential thrombocythemia myelofibrosis. J Clin Oncol. 2009;27(33):5587-5593.
doi pubmed - Morel P, Duhamel A, Hivert B, Stalniekiewicz L, Demory JL, Dupriez B. Identification during the follow-up of time-dependent prognostic factors for the competing risks of death and blast phase in primary myelofibrosis: a study of 172 patients. Blood. 2010;115(22):4350-4355.
doi pubmed - Gangat N, Wolanskyj AP, McClure RF, Li CY, Schwager S, Wu W, Tefferi A. Risk stratification for survival and leukemic transformation in essential thrombocythemia: a single institutional study of 605 patients. Leukemia. 2007;21(2):270-276.
doi pubmed - Skov V. Next generation sequencing in MPNs. Lessons from the past and prospects for use as predictors of prognosis and treatment responses. Cancers (Basel). 2020;12(8):2194.
doi pubmed - Cervantes F, Dupriez B, Pereira A, Passamonti F, Reilly JT, Morra E, Vannucchi AM, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113(13):2895-2901.
doi pubmed - Gangat N, Caramazza D, Vaidya R, George G, Begna K, Schwager S, Van Dyke D, et al. DIPSS plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. 2011;29(4):392-397.
doi pubmed - Tefferi A, Nicolosi M, Mudireddy M, Lasho TL, Gangat N, Begna KH, Hanson CA, et al. Revised cytogenetic risk stratification in primary myelofibrosis: analysis based on 1002 informative patients. Leukemia. 2018;32(5):1189-1199.
doi pubmed - Tefferi A, Jimma T, Gangat N, Vaidya R, Begna KH, Hanson CA, Van Dyke DL, et al. Predictors of greater than 80% 2-year mortality in primary myelofibrosis: a Mayo Clinic study of 884 karyotypically annotated patients. Blood. 2011;118(17):4595-4598.
doi pubmed - Quintas-Cardama A, Kantarjian H, Pierce S, Cortes J, Verstovsek S. Prognostic model to identify patients with myelofibrosis at the highest risk of transformation to acute myeloid leukemia. Clin Lymphoma Myeloma Leuk. 2013;13(3):315-318.e312.
doi pubmed - Vallapureddy RR, Mudireddy M, Penna D, Lasho TL, Finke CM, Hanson CA, Ketterling RP, et al. Leukemic transformation among 1306 patients with primary myelofibrosis: risk factors and development of a predictive model. Blood Cancer J. 2019;9(2):12.
doi pubmed - Grinfeld J, Nangalia J, Baxter EJ, Wedge DC, Angelopoulos N, Cantrill R, Godfrey AL, et al. Classification and personalized prognosis in myeloproliferative neoplasms. N Engl J Med. 2018;379(15):1416-1430.
doi pubmed - Harutyunyan A, Klampfl T, Cazzola M, Kralovics R. p53 lesions in leukemic transformation. N Engl J Med. 2011;364(5):488-490.
doi pubmed - Stegelmann F, Bullinger L, Schlenk RF, Paschka P, Griesshammer M, Blersch C, Kuhn S, et al. DNMT3A mutations in myeloproliferative neoplasms. Leukemia. 2011;25(7):1217-1219.
doi pubmed - Zhang SJ, Rampal R, Manshouri T, Patel J, Mensah N, Kayserian A, Hricik T, et al. Genetic analysis of patients with leukemic transformation of myeloproliferative neoplasms shows recurrent SRSF2 mutations that are associated with adverse outcome. Blood. 2012;119(19):4480-4485.
doi pubmed - Tefferi A, Lasho TL, Abdel-Wahab O, Guglielmelli P, Patel J, Caramazza D, Pieri L, et al. IDH1 and IDH2 mutation studies in 1473 patients with chronic-, fibrotic- or blast-phase essential thrombocythemia, polycythemia vera or myelofibrosis. Leukemia. 2010;24(7):1302-1309.
doi pubmed - Higgins A, Shah MV. Genetic and Genomic Landscape of Secondary and Therapy-Related Acute Myeloid Leukemia. Genes (Basel). 2020;11(7):749.
doi pubmed - Zhao LP, Cazaux M, Maslah N, De Oliveira RD, Verger E, Soret-Dulphy J, Marcault C, et al. Myeloproliferative Neoplasms (MPN) clonal evolution landscape and its impact on patients' prognosis. Blood. 2021;138(Supplement 1):317.
doi - Lundberg P, Karow A, Nienhold R, Looser R, Hao-Shen H, Nissen I, Girsberger S, et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood. 2014;123(14):2220-2228.
doi pubmed - Tsuruta-Kishino T, Koya J, Kataoka K, Narukawa K, Sumitomo Y, Kobayashi H, Sato T, et al. Loss of p53 induces leukemic transformation in a murine model of Jak2 V617F-driven polycythemia vera. Oncogene. 2017;36(23):3300-3311.
doi pubmed - Rampal R, Ahn J, Abdel-Wahab O, Nahas M, Wang K, Lipson D, Otto GA, et al. Genomic and functional analysis of leukemic transformation of myeloproliferative neoplasms. Proc Natl Acad Sci U S A. 2014;111(50):E5401-5410.
doi pubmed - Courtier F, Carbuccia N, Garnier S, Guille A, Adelaide J, Cervera N, Gelsi-Boyer V, et al. Genomic analysis of myeloproliferative neoplasms in chronic and acute phases. Haematologica. 2017;102(1):e11-e14.
doi pubmed - Mannelli F. Acute Myeloid Leukemia Evolving from Myeloproliferative Neoplasms: Many Sides of a Challenging Disease. J Clin Med. 2021;10(3):436.
doi pubmed - Mascarenhas J, Heaney ML, Najfeld V, Hexner E, Abdel-Wahab O, Rampal R, Ravandi F, et al. Proposed criteria for response assessment in patients treated in clinical trials for myeloproliferative neoplasms in blast phase (MPN-BP): formal recommendations from the post-myeloproliferative neoplasm acute myeloid leukemia consortium. Leuk Res. 2012;36(12):1500-1504.
doi pubmed - Tefferi A, Mudireddy M, Mannelli F, Begna KH, Patnaik MM, Hanson CA, Ketterling RP, et al. Blast phase myeloproliferative neoplasm: Mayo-AGIMM study of 410 patients from two separate cohorts. Leukemia. 2018;32(5):1200-1210.
doi pubmed - Kantharidis P, El-Osta A, deSilva M, Wall DM, Hu XF, Slater A, Nadalin G, et al. Altered methylation of the human MDR1 promoter is associated with acquired multidrug resistance. Clin Cancer Res. 1997;3(11):2025-2032.
- D'Altri T, Wilhelmson AS, Schuster MB, Wenzel A, Kalvisa A, Pundhir S, Meldgaard Hansen A, et al. The ASXL1-G643W variant accelerates the development of CEBPA mutant acute myeloid leukemia. Haematologica. 2021;106(4):1000-1007.
doi pubmed - Lancet JE, Uy GL, Newell LF, Lin TL, Ritchie EK, Stuart RK, Strickland SA, et al. CPX-351 versus 7+3 cytarabine and daunorubicin chemotherapy in older adults with newly diagnosed high-risk or secondary acute myeloid leukaemia: 5-year results of a randomised, open-label, multicentre, phase 3 trial. Lancet Haematol. 2021;8(7):e481-e491.
doi - Tam CS, Nussenzveig RM, Popat U, Bueso-Ramos CE, Thomas DA, Cortes JA, Champlin RE, et al. The natural history and treatment outcome of blast phase BCR-ABL- myeloproliferative neoplasms. Blood. 2008;112(5):1628-1637.
doi pubmed - Kennedy JA, Atenafu EG, Messner HA, Craddock KJ, Brandwein JM, Lipton JH, Minden MD, et al. Treatment outcomes following leukemic transformation in Philadelphia-negative myeloproliferative neoplasms. Blood. 2013;121(14):2725-2733.
doi pubmed - Badar T, Kantarjian HM, Ravandi F, Jabbour E, Borthakur G, Cortes JE, Pemmaraju N, et al. Therapeutic benefit of decitabine, a hypomethylating agent, in patients with high-risk primary myelofibrosis and myeloproliferative neoplasm in accelerated or blastic/acute myeloid leukemia phase. Leuk Res. 2015;39(9):950-956.
doi pubmed - Venton G, Courtier F, Charbonnier A, D'Incan E, Saillard C, Mohty B, Mozziconacci MJ, et al. Impact of gene mutations on treatment response and prognosis of acute myeloid leukemia secondary to myeloproliferative neoplasms. Am J Hematol. 2018;93(3):330-338.
doi pubmed - Gangat N, Guglielmelli P, Szuber N, Begna KH, Patnaik MM, Litzow MR, Al-Kali A, et al. Venetoclax with azacitidine or decitabine in blast-phase myeloproliferative neoplasm: A multicenter series of 32 consecutive cases. Am J Hematol. 2021;96(7):781-789.
doi pubmed - Castillo Tokumori F, Al Ali N, Chan O, Sallman D, Yun S, Sweet K, Padron E, et al. Comparison of different treatment strategies for blast-phase myeloproliferative neoplasms. Clin Lymphoma Myeloma Leuk. 2022;22(7):e521-e525.
doi pubmed - Thepot S, Itzykson R, Seegers V, Raffoux E, Quesnel B, Chait Y, Sorin L, et al. Treatment of progression of Philadelphia-negative myeloproliferative neoplasms to myelodysplastic syndrome or acute myeloid leukemia by azacitidine: a report on 54 cases on the behalf of the Groupe Francophone des Myelodysplasies (GFM). Blood. 2010;116(19):3735-3742.
doi pubmed - Andriani A, Montanaro M, Voso MT, Villiva N, Ciccone F, Andrizzi C, De Gregoris C, et al. Azacytidine for the treatment of retrospective analysis from the Gruppo Laziale for the study of Ph-negative MPN. Leuk Res. 2015;39(8):801-804.
doi pubmed - Mascarenhas JO, Rampal RK, Kosiorek HE, Bhave R, Hexner E, Wang ES, Gerds A, et al. Phase 2 study of ruxolitinib and decitabine in patients with myeloproliferative neoplasm in accelerated and blast phase. Blood Adv. 2020;4(20):5246-5256.
doi pubmed - Khan M, Siddiqi R, Gangat N. Therapeutic options for leukemic transformation in patients with myeloproliferative neoplasms. Leuk Res. 2017;63:78-84.
doi pubmed - Ruggiu M, Cassinat B, Kiladjian JJ, Raffoux E, Giraudier S, Robin M, Itzykson R, et al. Should Transplantation Still Be Considered for Ph1-Negative Myeloproliferative Neoplasms in Transformation? Biol Blood Marrow Transplant. 2020;26(6):1160-1170.
doi pubmed - Marcault C, Zhao LP, Maslah N, Verger E, Daltro de Oliveira R, Soret-Dulphy J, Cazaux M, et al. Impact of NFE2 mutations on AML transformation and overall survival in patients with myeloproliferative neoplasms. Blood. 2021;138(21):2142-2148.
doi pubmed - Harrison CN, Vannucchi AM, Kiladjian JJ, Al-Ali HK, Gisslinger H, Knoops L, Cervantes F, et al. Long-term efficacy and safety in COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for the treatment of myelofibrosis: 5-year final study results. Blood. 2015;126(23):59.
doi - Harrison CN, Vannucchi AM, Kiladjian JJ, Al-Ali HK, Gisslinger H, Knoops L, Cervantes F, et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia. 2016;30(8):1701-1707.
doi pubmed - Weber A, Borghouts C, Brendel C, Moriggl R, Delis N, Brill B, Vafaizadeh V, et al. Stat5 Exerts Distinct, Vital Functions in the Cytoplasm and Nucleus of Bcr-Abl+ K562 and Jak2(V617F)+ HEL Leukemia Cells. Cancers (Basel). 2015;7(1):503-537.
doi pubmed - Eghtedar A, Verstovsek S, Estrov Z, Burger J, Cortes J, Bivins C, Faderl S, et al. Phase 2 study of the JAK kinase inhibitor ruxolitinib in patients with refractory leukemias, including postmyeloproliferative neoplasm acute myeloid leukemia. Blood. 2012;119(20):4614-4618.
doi pubmed - Devillier R, Raffoux E, Rey J, Lengline E, Ronchetti AM, Sebert M, Boissel N, et al. Combination therapy with ruxolitinib plus intensive treatment strategy is feasible in patients with blast-phase myeloproliferative neoplasms. Br J Haematol. 2016;172(4):628-630.
doi pubmed - Talpaz M, Kiladjian JJ. Fedratinib, a newly approved treatment for patients with myeloproliferative neoplasm-associated myelofibrosis. Leukemia. 2021;35(1):1-17.
doi pubmed - Klener P, Sovilj D, Renesova N, Andera L. BH3 mimetics in hematologic malignancies. Int J Mol Sci. 2021;22(18):10157.
doi pubmed - Masarova L, DiNardo CD, Bose P, Pemmaraju N, Daver NG, Kadia TM, Chifotides HT, et al. Single-center experience with venetoclax combinations in patients with newly diagnosed and relapsed AML evolving from MPNs. Blood Adv. 2021;5(8):2156-2164.
doi pubmed - Konopleva M, Martinelli G, Daver N, Papayannidis C, Wei A, Higgins B, Ott M, et al. MDM2 inhibition: an important step forward in cancer therapy. Leukemia. 2020;34(11):2858-2874.
doi pubmed - Guo J, Roberts L, Chen Z, Merta PJ, Glaser KB, Shah OJ. JAK2V617F drives Mcl-1 expression and sensitizes hematologic cell lines to dual inhibition of JAK2 and Bcl-xL. PLoS One. 2015;10(3):e0114363.
doi pubmed - Harrison CN, Garcia JS, Somervaille TCP, Foran JM, Verstovsek S, Jamieson C, Mesa R, et al. Addition of navitoclax to ongoing ruxolitinib therapy for patients with myelofibrosis with progression or suboptimal response: phase II safety and efficacy. J Clin Oncol. 2022;40(15):1671-1680.
doi pubmed - Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL, Carney DA, et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol. 2012;30(5):488-496.
doi pubmed - McMurry H, Fletcher L, Traer E. IDH inhibitors in AML-promise and pitfalls. Curr Hematol Malig Rep. 2021;16(2):207-217.
doi pubmed - Tefferi A, Jimma T, Sulai NH, Lasho TL, Finke CM, Knudson RA, McClure RF, et al. IDH mutations in primary myelofibrosis predict leukemic transformation and shortened survival: clinical evidence for leukemogenic collaboration with JAK2V617F. Leukemia. 2012;26(3):475-480.
doi pubmed - Chifotides HT, Masarova L, Alfayez M, Daver N, Alvarado Y, Jabbour E, Konopleva M, et al. Outcome of patients with IDH1/2-mutated post-myeloproliferative neoplasm AML in the era of IDH inhibitors. Blood Adv. 2020;4(21):5336-5342.
doi pubmed - Padella A, Ghelli Luserna Di Rora A, Marconi G, Ghetti M, Martinelli G, Simonetti G. Targeting PARP proteins in acute leukemia: DNA damage response inhibition and therapeutic strategies. J Hematol Oncol. 2022;15(1):10.
doi pubmed - Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, Scott C, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376(9737):245-251.
doi - Zhao L, So CW. PARP-inhibitor-induced synthetic lethality for acute myeloid leukemia treatment. Exp Hematol. 2016;44(10):902-907.
doi pubmed - Pratz KW, Koh BD, Patel AG, Flatten KS, Poh W, Herman JG, Dilley R, et al. Poly (ADP-Ribose) polymerase inhibitor hypersensitivity in aggressive myeloproliferative neoplasms. Clin Cancer Res. 2016;22(15):3894-3902.
doi pubmed - Orta ML, Hoglund A, Calderon-Montano JM, Dominguez I, Burgos-Moron E, Visnes T, Pastor N, et al. The PARP inhibitor Olaparib disrupts base excision repair of 5-aza-2'-deoxycytidine lesions. Nucleic Acids Res. 2014;42(14):9108-9120.
doi pubmed - Patel PR, Senyuk V, Rodriguez NS, Oh AL, Bonetti E, Mahmud D, Barosi G, et al. Synergistic cytotoxic effect of busulfan and the PARP inhibitor veliparib in myeloproliferative neoplasms. Biol Blood Marrow Transplant. 2019;25(5):855-860.
doi pubmed - Nieborowska-Skorska M, Maifrede S, Dasgupta Y, Sullivan K, Flis S, Le BV, Solecka M, et al. Ruxolitinib-induced defects in DNA repair cause sensitivity to PARP inhibitors in myeloproliferative neoplasms. Blood. 2017;130(26):2848-2859.
doi pubmed - Abedin SM, Boddy CS, Munshi HG. BET inhibitors in the treatment of hematologic malignancies: current insights and future prospects. Onco Targets Ther. 2016;9:5943-5953.
doi pubmed - Sun Y, Han J, Wang Z, Li X, Sun Y, Hu Z. Safety and efficacy of bromodomain and extra-terminal inhibitors for the treatment of hematological malignancies and solid tumors: a systematic study of clinical trials. Front Pharmacol. 2020;11:621093.
doi pubmed - Dickinson M, Briones J, Herrera AF, Gonzalez-Barca E, Ghosh N, Cordoba R, Rutherford SC, et al. Phase 1b study of the BET protein inhibitor RO6870810 with venetoclax and rituximab in patients with diffuse large B-cell lymphoma. Blood Adv. 2021;5(22):4762-4770.
doi pubmed - Fiskus W, Sharma S, Qi J, Valenta JA, Schaub LJ, Shah B, Peth K, et al. Highly active combination of BRD4 antagonist and histone deacetylase inhibitor against human acute myelogenous leukemia cells. Mol Cancer Ther. 2014;13(5):1142-1154.
doi pubmed - Braun T, Gardin C. Investigational BET bromodomain protein inhibitors in early stage clinical trials for acute myelogenous leukemia (AML). Expert Opin Investig Drugs. 2017;26(7):803-811.
doi pubmed - Saenz DT, Fiskus W, Manshouri T, Rajapakshe K, Krieger S, Sun B, Mill CP, et al. BET protein bromodomain inhibitor-based combinations are highly active against post-myeloproliferative neoplasm secondary AML cells. Leukemia. 2017;31(3):678-687.
doi pubmed - Eskandari M, Shi Y, Liu J, Albanese J, Goel S, Verma A, Wang Y. The expression of MDM2, MDM4, p53 and p21 in myeloid neoplasms and the effect of MDM2/MDM4 dual inhibitor. Leuk Lymphoma. 2021;62(1):167-175.
doi pubmed - Toledo F, Wahl GM. MDM2 and MDM4: p53 regulators as targets in anticancer therapy. Int J Biochem Cell Biol. 2007;39(7-8):1476-1482.
doi pubmed - Latif AL, Newcombe A, Li S, Gilroy K, Robertson NA, Lei X, Stewart HJS, et al. BRD4-mediated repression of p53 is a target for combination therapy in AML. Nat Commun. 2021;12(1):241.
doi pubmed - Wang HQ, Mulford IJ, Sharp F, Liang J, Kurtulus S, Trabucco G, Quinn DS, et al. Inhibition of MDM2 promotes antitumor responses in p53 wild-type cancer cells through their interaction with the immune and stromal microenvironment. Cancer Res. 2021;81(11):3079-3091.
doi pubmed - Andreeff M, Kelly KR, Yee K, Assouline S, Strair R, Popplewell L, Bowen D, et al. Results of the phase I trial of RG7112, a small-molecule MDM2 antagonist in leukemia. Clin Cancer Res. 2016;22(4):868-876.
doi pubmed - Stein EM, DeAngelo DJ, Chromik J, Chatterjee M, Bauer S, Lin CC, Suarez C, et al. Results from a first-in-human phase I study of siremadlin (HDM201) in patients with advanced wild-type TP53 solid tumors and acute leukemia. Clin Cancer Res. 2022;28(5):870-881.
doi pubmed - Konopleva MY, Rollig C, Cavenagh J, Deeren D, Girshova L, Krauter J, Martinelli G, et al. Idasanutlin plus cytarabine in relapsed or refractory acute myeloid leukemia: results of the MIRROS trial. Blood Adv. 2022;6(14):4147-4156.
doi pubmed - Kubesova B, Pavlova S, Malcikova J, Kabathova J, Radova L, Tom N, Tichy B, et al. Low-burden TP53 mutations in chronic phase of myeloproliferative neoplasms: association with age, hydroxyurea administration, disease type and JAK2 mutational status. Leukemia. 2018;32(2):450-461.
doi pubmed - Marcellino BK, Farnoud N, Cassinat B, Lu M, Verger E, McGovern E, Patel M, et al. Transient expansion of TP53 mutated clones in polycythemia vera patients treated with idasanutlin. Blood Adv. 2020;4(22):5735-5744.
doi pubmed - Sallman DA, DeZern AE, Garcia-Manero G, Steensma DP, Roboz GJ, Sekeres MA, Cluzeau T, et al. Eprenetapopt (APR-246) and azacitidine in TP53-mutant myelodysplastic syndromes. J Clin Oncol. 2021;39(14):1584-1594.
doi pubmed - Roskoski R, Jr. Cyclin-dependent protein serine/threonine kinase inhibitors as anticancer drugs. Pharmacol Res. 2019;139:471-488.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.