

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://www.thejh.org |
Case Report
Volume 12, Number 1, February 2023, pages 49-58

Transformed Plasmablastic Lymphoma Presenting With Marked Lymphocytosis and Spontaneous Tumor Lysis Syndrome
Yannis Hadjiyannisa, d ![]() , Cecelia Millerb, Norris I. Hollieb, Jayalakshmi Balakrishnab, Francesca Cottinic, d
, Cecelia Millerb, Norris I. Hollieb, Jayalakshmi Balakrishnab, Francesca Cottinic, d ![]()
aDepartment of Pathology, University of Pittsburgh Medical Center, Pittsburgh, PA 15261, USA
bDepartment of Pathology, Wexner Medical Center, The Ohio State University, Columbus, Columbus, OH 43201, USA
cDivision of Hematology, Department of Internal Medicine, The Ohio State University Comprehensive Cancer Center, Columbus, OH 43201, USA
dCorresponding Author: Francesca Cottini, Division of Hematology, Department of Internal Medicine, College of Medicine, The Ohio State University, Columbus, OH 43210, USAand Yannis Hadjiyannis, Department of Pathology, University of Pittsburgh Medical Center, Pittsburgh, PA 15261, USA
Manuscript submitted December 15, 2022, accepted February 6, 2023, published online February 25, 2023
Short title: Transformed Complex Plasmablastic Lymphoma
doi: https://doi.org/10.14740/jh1067
| Abstract | ▴Top |
The clinicopathology entity of plasmablastic lymphoma (PBL), despite broad recognition by the World Health Organization (WHO), represents a diagnostic challenge due to its overlapping features and scarce occurrence. Often, PBL arises in immunodeficient, elderly male patients, most notably those who are human immunodeficiency virus (HIV)-positive. More infrequent, cases of transformed PBL (tPBL) evolved from another hematologic disease have been identified. Herein, we describe a case of a 65-year-old male transferred from a neighboring hospital with pronounced lymphocytosis and spontaneous tumor lysis syndrome (sTLS) presumed to be chronic lymphocytic leukemia (CLL). Utilizing a complete clinical, morphologic, immunophenotypic, and molecular evaluation, we arrived at a final diagnosis of tPBL with sTLS, suspected to have evolved from the NF-κB/NOTCH/KLF2 (NNK) genetic cluster of splenic marginal zone lymphoma (SMZL) (NNK-SMZL), a potential transformation and presentation, to our knowledge, not previously reported. However, definitive clonality testing was not performed. In this report, we also outline the diagnostic and educational considerations we faced in discerning tPBL from other more common B-cell malignancies which can present similarly, such as CLL, mantle cell lymphoma, or plasmablastic myeloma. We summarize recently reported molecular, prognostic, and therapeutic considerations for the treatment and recognition of PBL, including the successful implementation, in our patient, of bortezomib to an EPOCH (etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin) regimen with prophylactic intrathecal methotrexate, who has since achieved complete remission (CR) and entered clinical surveillance. Lastly, this report briefly highlights the challenge we faced in this area of hematologic typification that necessitates additional review and discussion by the WHO: tPBL with potential double-hit cytogenetic versus double-hit lymphoma with a plasmablastic phenotype.
Keywords: Plasmablastic lymphoma; Splenic marginal zone lymphoma; EBV; Tumor lysis syndrome; Double-hit lymphoma
| Introduction | ▴Top |
A decade after its initial description, the clinicopathologic entity of primary plasmablastic lymphoma (PBL) was recognized by the World Health Organization (WHO) in 2017 as a subtype of lymphoma [1]. Despite broad recognition, PBL often represents a diagnostic challenge due to its infrequent occurrence and overlapping features. Usually, PBL occurs in elderly male patients with underlying immunodeficiencies, most notably those who are human immunodeficiency virus (HIV)-positive [1-4]. In a minority of cases, transformed PBL (tPBL) arises from a preexisting malignancy, such as chronic lymphocytic leukemia (CLL) or follicular lymphoma [2, 5, 6], but it has never been reported as a potential transformation from splenic marginal zone lymphoma (SMZL). Patients often present in stage III/IV with various involved sites depending on clinical factors such as HIV and Epstein-Barr virus (EBV) status [2, 4, 7].
Cases of primary or de novo PBL are characterized by an aggressive, cluster of differentiation (CD)20-negative, plasmablastic phenotype (CD38, CD138, melanoma associated antigen (mutated) 1 (MUM1)-positive) with Myc proto-oncogene protein (MYC) rearrangements and high genetic complexity. In cases of tPBL, the cytogenetic aberrations are often more complicated. MYC rearrangements are still a diagnostic hallmark, but other concurrent abnormalities, such as B-cell lymphoma 2 (BCL2) rearrangements, may be present as seen in reported cases of tPBL arising from follicular lymphoma or diffuse large B-cell lymphoma (DLBCL) [5, 8]. Unfortunately, the presence of these two cytogenetic aberrations cast further challenge on the diagnosis of primary or tPBL as the WHO has yet to address whether these cases represent a high-grade double-hit lymphoma (DHL) with a plasmablastic phenotype or primary or tPBL with double-hit cytogenetics. Concurrent and temporal cytogenetics studies are likely required, capturing just a minority of these cases.
Further, no standard therapeutic regimen exists due to the rarity of de novo and tPBL. Recent retrospective studies have demonstrated that the addition of bortezomib either to lymphoma or acute lymphoblastic leukemia protocol is effective [9]. However, continued therapeutic study, implementation, and review is needed.
| Case Report | ▴Top |
Investigations
A 65-year-old male patient with no significant past medical history presented, as a transfer, for evaluation of prolonged lymphocytosis (> 2 months) presumed by the outside institution to be CLL with spontaneous tumor lysis syndrome (sTLS). At the neighboring hospital, the patient was admitted for severe diarrhea, abdominal pain, and hyperuricemia (14.0 mg/dL) with acute kidney injury (AKI) (Cr 1.86 mg). His clinical course was complicated by grade IV esophagitis with coffee-ground emesis, recent incarcerated hernia repair, vancomycin-resistant enterococci bacteremia, and rotavirus gastroenteritis. Computed tomography (CT) demonstrated abdominal wall edema, diffuse bowel thickening, retroperitoneal adenopathy, and an enlarged spleen (15.8 cm) with a 3.8 cm splenic infarct. Upon transfer for hematologic evaluation, his admission labs revealed leukocytosis (30.10 × 103/µL), warm autoimmune hemolytic anemia (hemoglobin (Hgb) 9.9 g/dL, direct antiglobulin test (DAT) C3, immunoglobulin G (IgG)-positive with a warm autoantibody), hyperkalemia (5.2 mmol/L), hyperuricemia (11.3 mg/dL), and elevated lactate dehydrogenase (LDH, 2,673 U/L). Corrected calcium and phosphate were within normal limits, and resolution of AKI occurred following two doses of rasburicase and bolus hydration.
Following the resolution of his AKI, normal saline (125 mL/h) and allopurinol were initiated, with stabilization of his serum creatinine (1.24 mg/dL) and hyperuricemia (6.6 mg/dL). Peripheral blood smear revealed a lymphocytosis (26.49 × 103/µL) of small- to medium-sized lymphocytes with round nuclei and clumped chromatin, frequent smudge or basket cells, moderate anemia with anisocytosis and polychromasia, and thrombocytosis with unremarkable platelet morphology (Fig. 1a). Peripheral blood immunophenotyping by flow cytometry demonstrated a population of monotypic, kappa light chain restricted B lymphocytes that were CD19-positive, CD20-moderate, CD5, CD10 and CD23-negative (Fig. 1b). Positron emission tomography-computed tomography (PET-CT) scan demonstrated diffuse, hypermetabolic foci in the spleen, bilateral lower lobes of the lungs, mediastinal pleura, anorectal region, peritoneal cavity, bowel loop, and pelvic wall compatible with malignant involvement (Fig. 2). No focal, hypermetabolic osseous lesions were identified (Fig. 2). Shortly after, the patient became febrile, tachycardic, and his severe diarrhea reoccurred with diffuse abdominal pain. Repeat CT demonstrated diffuse colonic thickening suggestive of infectious colitis, and stool culture confirmed Clostridium difficile infection requiring initiation of oral vancomycin with the resolution of his symptoms.
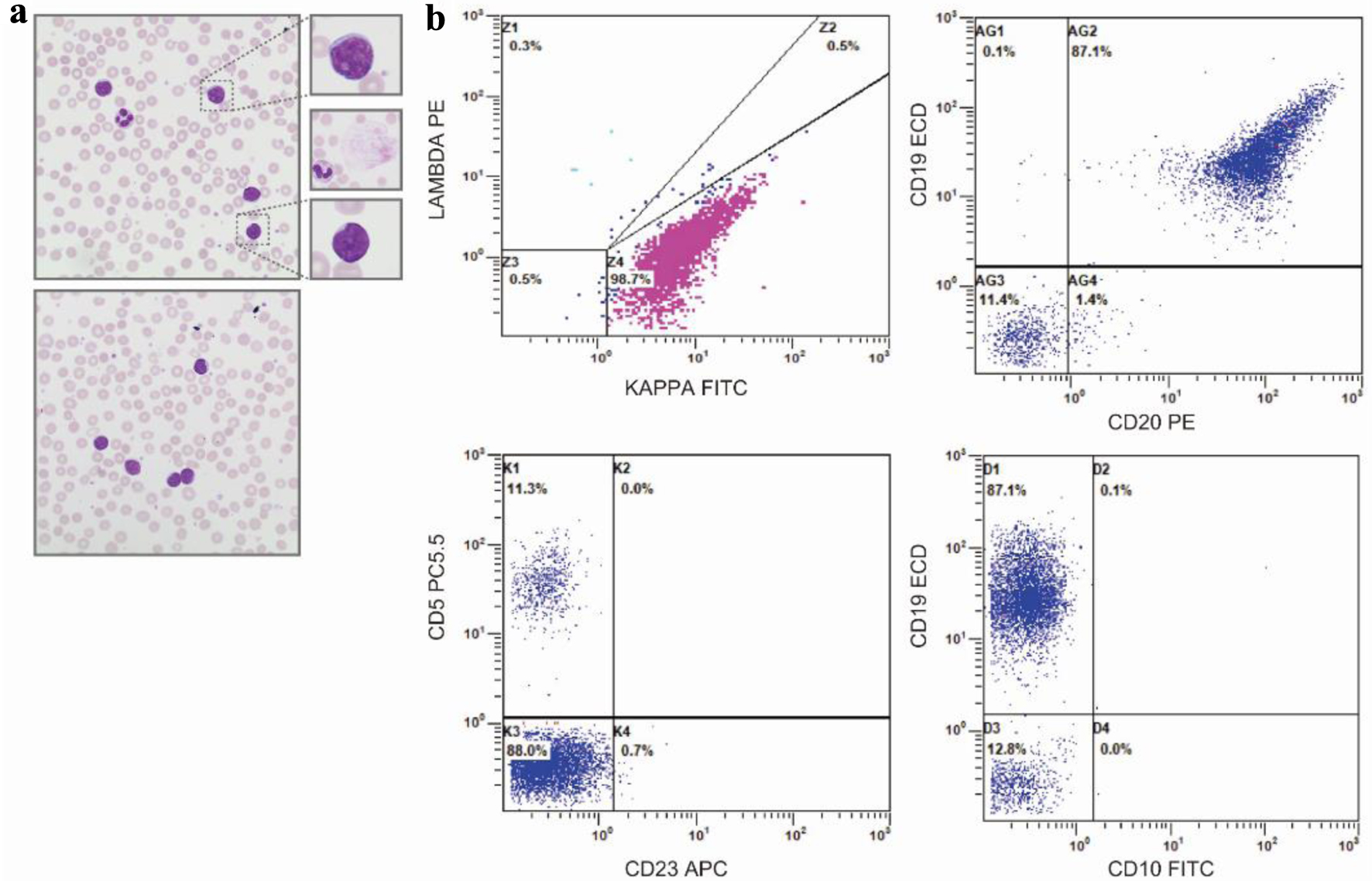 Click for large image | Figure 1. Peripheral blood smear and flow cytometry. (a) Representative, × 50 oil immersion microscopy of the peripheral blood smear - with digitally enlarged × 10 insets - demonstrating small- to moderate-sized lymphoid cells with scant to moderate cytoplasm, round to slightly irregular nuclei, and clumped chromatin. Middle inset, representative smudge, or basket cell present throughout smear. (b) Peripheral blood immunophenotype by flow cytometry demonstrating a monoclonal kappa B-cell population (top, left plot), with strong expression of CD19, moderate expression of CD20 (top, right panel), negative expression of CD5, and negative expression of CD23 (bottom, left panel) and CD10 (bottom, right panel). |
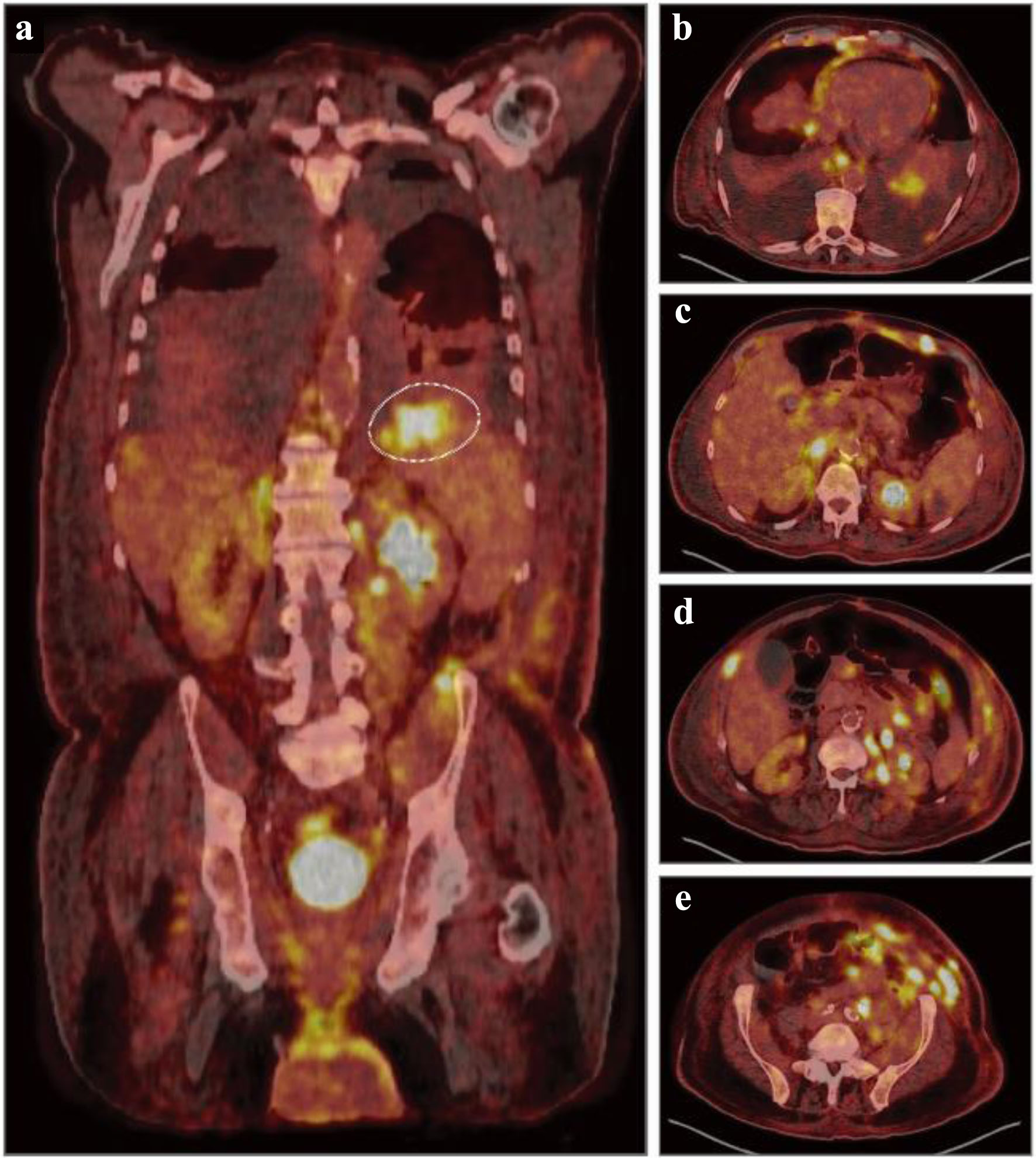 Click for large image | Figure 2. Diagnostic, fluorodeoxyglucose (FDG) positron emission tomography combined with computed tomography (PET-CT). (a) Representative coronal PET-CT image demonstrating diffuse uptake and hypermetabolic foci throughout the liver, spleen, kidneys, bowel, left iliac chain and inguinal region concerning for malignant involvement. Dashed circle indicating a hypermetabolic focus with a maximum standardized uptake value (SUV) of 10.0 in the superior splenic pole. (b-e) Representative axial, nonsequential PET-CT images demonstrating numerous hypermetabolic foci throughout the bilateral lower lobes, mediastinal pleura, esophagus, liver, kidney, abdominal wall, peritoneal cavity, and pelvic wall concerning for malignant involvement. |
On bone marrow (BM) biopsy, nodular and interstitial lymphoid (20-30% CD3 T cells; 20% paired box transcription factor 5 (PAX5)/CD20 B cell) infiltrates comprised of small cells with mature-appearing chromatin were evident (Fig. 3). Cyclin D1 and SRY-related HMG-box transcription factor 11 (SOX11) were negative, ruling out mantle cell lymphoma. No overt hemophagocytic histiocytes were present (Fig. 3). Immunophenotypic analysis of the BM demonstrated a monoclonal, kappa population (44.7% of events, 84.5% of lymphocytes) of B lymphocytes that express CD20 (moderate to bright), CD19, CD38, FMC7, CD22 with a subset expressing CD79b, and negative for CD10, CD23, CD5, CD43, CD9, CD123, CD11c, CD25, and CD103, ruling out hairy cell leukemia. Fluorescence in situ hybridization (FISH) studies of the BM aspirate were positive for MYC and BCL2 rearrangements (Table 1). BM aspirate cytogenetics displayed a complex karyotype with an interstitial q-arm deletion on chromosome 7, a three-way translocation between chromosomes 8, 14, and 18 - resulting in immunoglobulin heavy chain (IGH)-MYC and IGH-BCL2 rearrangements - and a translocation between chromosomes 9 and 14 (Table 1). DNA extracted from sorted CD19-positive B cells for next-generation sequencing (NGS) analysis revealed dual mutations in Kruppel-like factor 2 (KLF2), a zinc finger transcription factor - including one to our knowledge not previously reported - but no mutations in caspase recruitment domain-containing protein 11 (CARD11), myeloid differentiation primary response 88 (MYD88), CD79A/B, C-X-C chemokine receptor type 4 (CXCR4), or CLL-associated genes (Table 1).
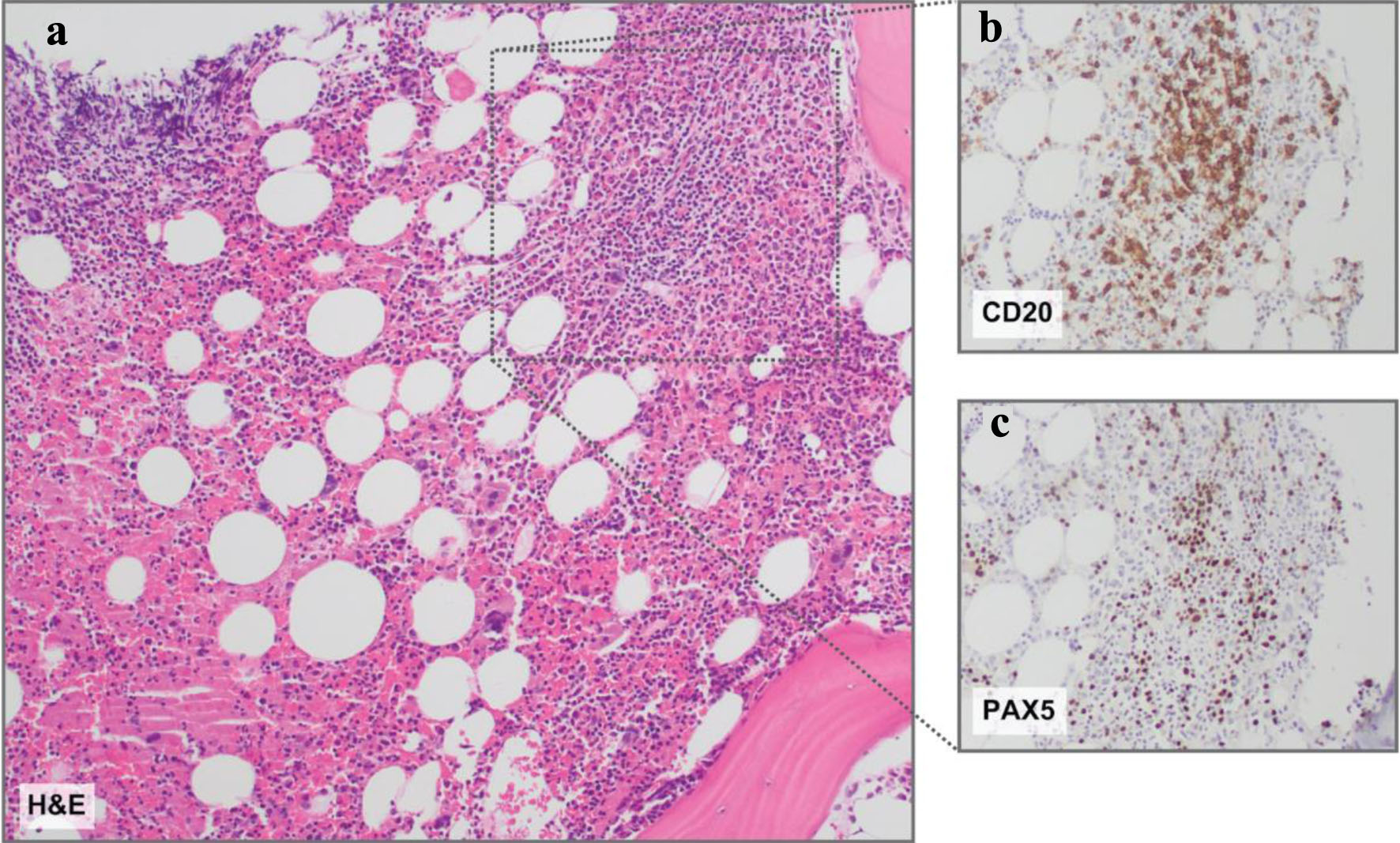 Click for large image | Figure 3. Bone marrow histopathology. (a) Representative hematoxylin and eosin (H&E) stain revealing a slightly nodular and interstitial lymphoid infiltrate comprised of small lymphoid cells. (b) CD20 and (c) PAX5 immunohistochemistry insets demonstrating B-cell antigens on the nodular and interstitial lymphoid infiltrate comprised of small lymphoid cells. Magnification: all, × 10. |
 Click to view | Table 1. Cytogenetics and Next-Generation Sequencing Results |
Core needle biopsy of the abdominal wall nodule demonstrated hematolymphoid infiltrate with anaplastic features (Fig. 4). Large, pleomorphic CD20-negative, CD45-positive, kappa-restricted cells with prominent nucleoli expressing MUM1, CD30, CD138, and MYC were identified with a proliferative rate shown by Ki-67 of > 90% (Fig. 4). Human gamma herpesvirus 8 (HHV-8), anaplastic lymphoma kinase (ALK), BCL2, and B-cell lymphoma 6 (BCL6) immunohistochemistries, and Epstein-Barr encoding region (EBER)-in situ hybridization (ISH) were negative. The nodule biopsy was positive for MYC rearrangement, equivocal for BCL2 due to tumor necrosis, and negative for BCL6 rearrangements (Table 1).
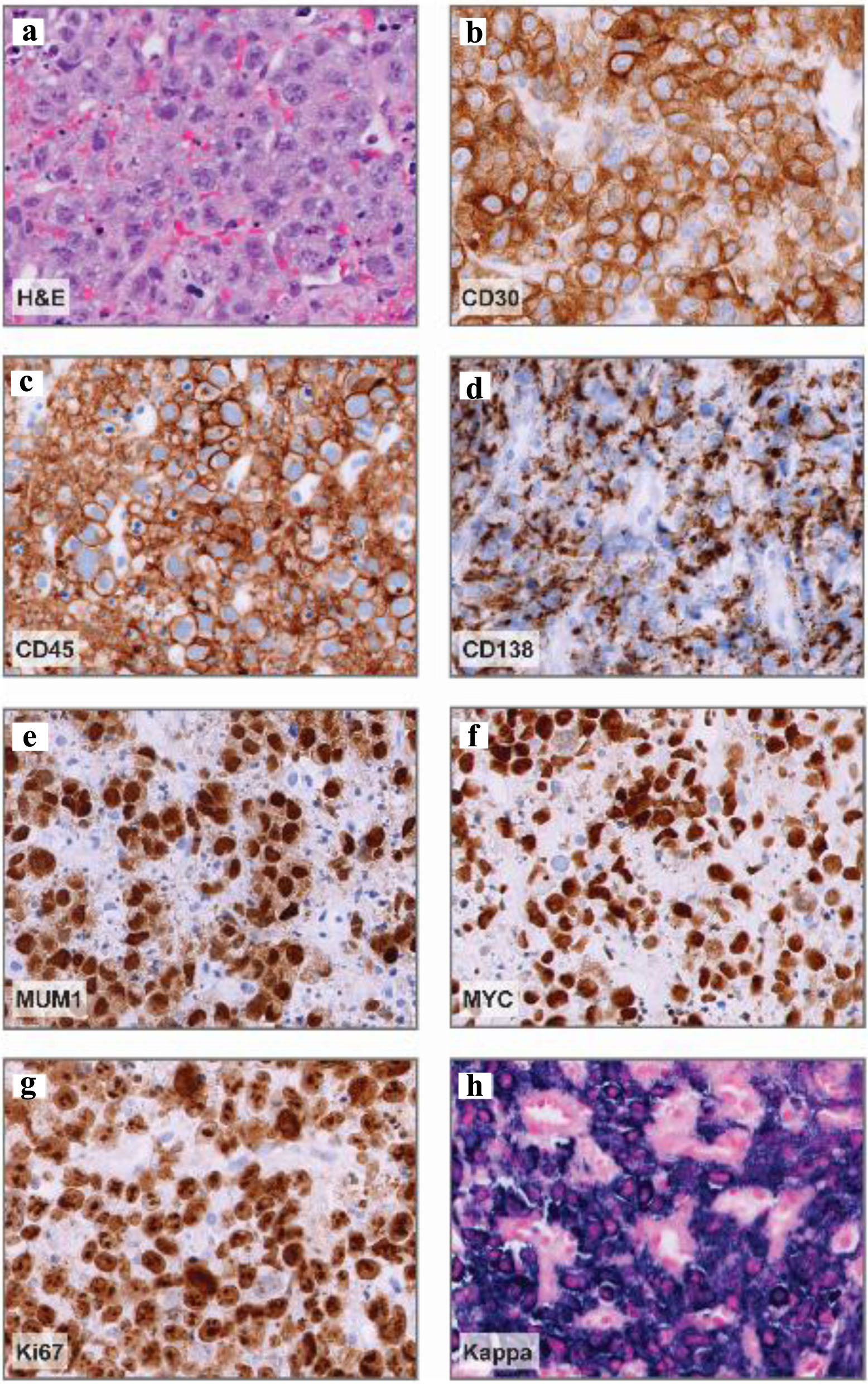 Click for large image | Figure 4. Abdominal nodule histopathology. (a) Representative hematoxylin and eosin (H&E) stain demonstrating large, pleomorphic cells with prominent nucleoli and vesicular chromatin. (b-e) Representative CD30, CD45, CD138, and MUM1 immunohistochemistry images demonstrating positive staining in a vast majority of neoplastic cells (about 95%) and variable CD138 expression. (f) MYC immunohistochemistry demonstrating diffuse expression in neoplastic cells. (g) Ki-67 immunohistochemistry, proliferation index: > 90%. (h) Kappa in situ hybridization denoting kappa-restricted, neoplastic cells. Magnification: all, × 40. |
No monoclonal protein was detected in the serum; elevated serum kappa free light chains (49.6 mg/L) with a mildly increased kappa-to-lambda ratio of 2.64 (reference range: 0.51 - 1.72) were observed. Monoclonal free kappa light chains were detected in the urine. Cytomegalovirus (CMV) (< 50 IU/mL), HHV-8 (< 1,000 copies/mL), and EBV polymerase chain reaction (PCR) (< 1,000 IU/mL) were negative. Serologic HIV evaluation was non-reactive and hepatitis serologies were negative. Ferritin and triglycerides were slightly elevated at 1,492.7 ng/mL and 150 mg/dL, respectively. Aspartate aminotransferase was within normal limits (32 U/L). Utilizing the weighted criteria of the hemophagocytic lymphohistiocytosis (HLH)-probability score (HScore), the calculated probability of acquired malignancy-associated HLH was low (< 1%, 86 points (optimal cutoff: 169 points, sensitivity: 93%, specificity, 86%)) [10]. Brain magnetic resonance imaging (MRI) demonstrated no abnormal parenchymal enhancement or intracranial abnormalities; no abnormalities or malignant cells were evident on cerebrospinal fluid (CSF) analysis.
Diagnosis
The immunohistochemistry, immunophenotyping, and NGS studies ruled out CLL, mantle cell lymphoma, hairy cell leukemia, and HHV8-driven lymphomas. Plasmablastic myeloma was excluded, given the absence of a serum monoclonal protein, the free kappa-to-lambda ratio less than 100, and the lack of osseous lesions with a normal calcium level. The presence of splenomegaly in addition to the 18F-fluorodeoxyglucose (FDG) uptake in the superior splenic pole by PET-CT, with a standardized uptake value (SUV) of 10, suggested a diagnosis of SMZL.
Despite a lack of intrasinusoidal BM involvement commonly seen in but not specific for SMZL, the immunohistochemistry was positive for CD19, CD20, CD79b and negative for CD5 and CD23 congruent with SMZL [11, 12]. Moreover, the presence of a similar kappa monoclonal B-cell population (CD19, CD20 positive, CD5, CD10, and CD23 negative) by flow cytometry in the peripheral blood and BM suggested SMZL with BM and peripheral blood involvement [13-16]. The KLF2 mutations on sorted CD19-positive B cells and the loss of chromosome 7q also advocated for SMZL [13-16]. Indeed, KLF2 mutations have rarely been reported in cases of other lymphomas, and almost exclusively occur in the presence of BCL6 rearrangements or a BCL6 signature [17].
Nonetheless, the clinical history, laboratory results, presence of extranodal sites of disease by PET-CT, and identification of a complex karyotype with translocations in MYC, and BLC2 suggested underlying SMZL transformation observed in roughly about 20% of patients with SMZL with a rapidly progressive course [12, 13]. The abdominal nodule biopsy demonstrated large, pleomorphic CD20-negative and MUM1, CD30, MYC, and CD138-positive cells favoring a potential diagnosis of PBL [1]. The negative infectious evaluation and EBER-ISH on the abdominal nodule biopsy pointed away from several underlying etiologies of primary or de novo PBL [1-4, 7, 11] and towards a potential transformation.
A final possibility was a DHL with a plasmablastic phenotype [18]. While the initial evaluation demonstrated MYC rearrangements and discrepant BCL2 results between the BM and nodule biopsy, work by Kim et al demonstrated the potential for discordant FISH results between BM aspirate and extranodal or nodal tissue arguing against double-hit cytogenetics in the abdominal nodule [18]. Further, cases of DHL are significantly associated with a CD5-negative and CD10-positive immunophenotype, whereas both our peripheral and BM aspirate immunophenotypic assessment revealed a CD5/CD10-negative characteristic pointing away from the diagnosis of DHL with a plasmablastic phenotype [18] and towards tPBL with complex cytogenetics.
Although we could not definitively prove a clonal relationship between the two aforementioned components, the final diagnosis was determined to be Ann Arbor stage IV tPBL, likely arising from SMZL with peripheral blood and BM involvement. The revised international prognostic index (R-IPI) score and the central nervous system (CNS)-IPI score was 4. The calculated 2-year risk of CNS progression/recurrence was 10.2%, and the National Comprehensive Cancer Network IPI (NCCN-IPI) score was 6, further accentuating the aggressive nature of the patient’s disease.
Treatment
Given the aggressive nature and rare occurrence of PBL, six cycles of V-EPOCH (bortezomib, etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin) every 21 days with intrathecal methotrexate for the first four cycles were initiated. V-EPOCH was administered in the standard approach, consisting of a 96-h continuous infusion of etoposide IV, doxorubicin IV, and vincristine followed by cyclophosphamide IV on day 5. Bortezomib was given at a dose of 1.3 mg/m2 on days 1 and 4 of each cycle. For our patient, the main side effects were diarrhea and neutropenia. Limiting toxicity was neuropathy, resulting in dose-reduction and then discontinuation of vincristine. The treatment of PBL still remains controversial.
Given the rarity of the disease, no specific prospective studies are available and patients with PBL are often excluded from clinical trials. Makady et al presented a meta-analysis of 173 patients reported in the literature and treated for PBL, with 24 of them receiving V-EPOCH (eight patients in Dittus et al and 16 patients in Castillo et al). Of this small cohort, 11/24 patients were HIV-positive and none had tPBL. All 24 patients received V-EPOCH as first line of therapy for 2 - 6 cycles; 23/24 had a CR; 4/24 underwent autologous stem cell transplant in first remission. Five out of eight patients of Dr. Dittus’s study received methotrexate intrathecally or high-dose methotrexate for proven CNS disease or CNS prophylaxis, while CNS treatment or prophylaxis was not reported in Dr. Castillo’s study.
The median follow-up was 4 years, with a 2-year progression-free survival and overall survival (OS) of 50% in Dr. Dittus’s study and a median OS of 62 months, with a 5-year OS rate of 63% in Dr. Castillo’s study. In term of toxicities, grade 3 or higher adverse events included thrombocytopenia (7/24, 29.1%), febrile neutropenia (6/24, 25%), and neuropathy (7/24, 29.1%). Despite the low patient numbers, the addition of bortezomib seemed to increase the risk of neuropathy which otherwise occurs in 10-18.6% of patients treated with regular EPOCH [9, 19, 20].
Follow-up and outcomes
After six cycles of V-EPOCH, end of therapy PET-CT indicated CR, and clinical surveillance every 3 months was initiated. Spleen size also normalized to 11.5 cm. The patient tolerated the chemotherapy regimen and is experiencing ongoing remission.
| Discussion | ▴Top |
A hallmark of PBL is the plasma cell-like phenotype or malignant plasmablast [2]. Conventionally, a plasmablast is a short-lived, immature cell type, a part of the plasma cell lineage. While the exact pathogenesis of PBL remains elusive, several genetic, infectious, and immunologic perturbations have been purposed to affect the maturation, transformation, coordination, and survival of plasmablasts such as EBV antigen-driven apoptotic survival, MYC dysregulation, or genetic alterations affecting the receptor tyrosine kinase-rat sarcoma virus-rapidly accelerated fibrosarcoma (RTK-RAS-RAF), Janus kinase-signal transducer activator of transcription (JAK-STAT), induced myeloid leukemia cell differentiation protein (MCL1), interferon regulatory factor 4 (IRF4), mitogen-activated protein kinase (MAPK), and NOTCH pathways [21-23].
Unique oncogenic mutations and a differential set of expression patterns have also been reported in EBV-negative versus EBV-positive cases [24]. EBV-negative cases of PBL had a higher mutational and copy-number load as well as more frequent tumor protein 53 (TP53), CARD11, and MYC mutations [21, 22]. Whereas EBV-positive cases had more frequent mutations in the JAK-STAT pathway [21, 22]. Interestingly, PBL usually represses the B-cell receptor (BCR) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling [23, 25], while KLF2 inactivating mutations, like the ones present in the BM sample of our patient, have the opposite effect, allowing for NF-κB activation by BCR [14].
Further, genetic alterations in KLF2 underscore one of the prominent genetic clusters of SMZL termed NNK-SMZL [13]. Within this genetic cluster, KLF2 mutations were enriched in the immune-suppressive subtype of NNK-SMZL, which had a heavier neoantigen load and inflammatory tumor microenvironment [13]. Since data from the abdominal nodule, to evaluate for clonality and persistence of the KLF2 mutation, are not available, we can only speculate that the more aggressive and proliferative nature of NNK-SMZL with immune-suppressive features might have created an environment suitable for plasmablastic transformation with complex cytogenetics and clonal independence from NF-κB signaling.
In our case of Ann Arbor stage IV tPBL, the patient met three of the four criteria for sTLS [24]: uric acid ≥ 8 mg/dL, suspected malignancy with elevated LDH more than twice the upper limit, and acute non-postobstructive oliguric kidney injury. Uc acid level was not collected; thus, the uric acid to creatinine ratio > 1 was not evaluated. Moreover, the lack of hyperphosphatemia, a common occurrence in sTLS, suggested the re-utilization of free phosphorus by rapidly proliferating malignant cells. To our knowledge, sTLS has yet to be reported in cases of PBL.
Generally, the prognosis of PBL is considered dismal, with a median OS of approximately 8 months [3]. Some patients have better outcomes, such as those with disease sites involving the head/neck region or the immunocompetent with EBV-positive disease. In contrast, a high IPI score, Ki-67 expression of > 80%, and TP53 mutations have been associated with worse OS [9]. A retrospective, multicenter evaluation of the IPI, R-IPI, and the NCCN-IPI indexes also supported their use in patients with PBL for their prognostic capabilities [26]. Validation of the CNS-IPI score in patients with PBL has not been examined and CNS prophylaxis with intrathecal methotrexate is largely controversial and based on institutional practices. Additional studies on the use of CNS-IPI and CNS prophylaxis in PBL are thus warranted.
Given the rarity of PBL, clinical trials to define the best treatment approach are limited. Systematic retrospective analyses revealed promising results upon the addition of the proteasome inhibitor, bortezomib, to either lymphoma or acute lymphoblastic leukemia regimens [9]. For instance, V-EPOCH regimens obtained a CR rate of 90-100%, reduced relapse rates, and demonstrated a 5-year OS rate of 65% in patients with PBL [9]. Indeed, despite poor prognostic features, such as EBV-negativity, a Ki-67 index > 80%, and high-risk IPI-R/CNS-IPI/NCCN-IPI scores, our patient benefited from the addition of bortezomib to an EPOCH regimen with prophylactic intrathecal therapy, having achieved ongoing remission.
Finally, given the plasmablastic phenotype with CD138- and CD38-positivity, other therapies used in patients with multiple myeloma have been implemented. Lenalidomide in combination with cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) [26, 27] or daratumumab (monoclonal antibody against CD38) in combination with EPOCH demonstrated responses in selected patients with advanced PBL [28]. In relapsed or refractory cases of aggressive lymphoma, therapeutic options have also expanded to incorporate experimental immunotherapy. As such, Castillo et al demonstrated an effective response in the first case of refractory, recurrent PBL treated with the anti-PD1 monoclonal antibody, pembrolizumab [29]. Moreover, promising therapeutically actionable pathways are present in PBL, such as the JAK-STAT and the RTK-RAS pathways, including a newly reported neurotrophic tyrosine kinase receptor type 3 (NTRK3) activating mutation in a minor subset of PBL cases [21-23]. Ultimately, alterations in these pathways suggest a role for JAK-STAT and pan-TRK inhibitors. In vitro models of JAK-STAT inhibition with tofacitinib demonstrated encouraging results [21], but neither JAK-STAT nor pan-TRK inhibitors have seen clinical implementation, and further study is required before broad implementation. In our case, such therapeutic options were not considered as molecular alterations in these specific pathways were not assessed.
Conclusion
In sum, we present, to our knowledge, a case of tPBL with sTLS suspected to arise from NNK-SMZL. While we cannot prove temporal and clonal transforation, our report highlights the importance of the differential and the effect of minimizing anchoring bias and heuristics given the patient’s presumptive diagnosis and complex presentation. It underscores the significance of a complete clinical, morphologic, immunophenotypic, and molecular analysis as part of hematologic typification. It illuminates and summarizes unique diagnostic and therapeutic considerations for the recognition and treatment of this exceedingly rare entity. Lastly, it raises several scientific, clinical, and therapeutic challenges and future directions associated with the typification and treatment of PBL.
Learning points
This case provides several learning points associated with the etiology, therapeutic considerations, and study of PBL: first, it reports on the diagnostic criteria and identification of sTLS and demonstrates its efficacy in PBL; second, it demonstrates the unique diagnostic considerations associated with PBL versus several other conditions; third, it reviews recent molecular, therapeutic, and prognostic discovers vital to the typification and treatment of patients with PBL. Finally, it illustrates the challenge and continued need for further study of PBL.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
Informed consent has been obtained.
Author Contributions
Dr. Hadjiyannis and Dr. Cottini provided patient care. Dr. Hadjiyannis, Cottini, Balakrishna, Hollie, and Miller accomplished the acquisition of all clinical data. The report was drafted by Dr. Hadjiyannis and revised by all authors. All authors approved the final submitted manuscript.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
Abbreviations
PBL: plasmablastic lymphoma; WHO: World Health Organization; tPBL: transformed plasmablastic lymphoma; sTLS: spontaneous tumor lysis syndrome; CLL: chronic lymphocytic leukemia; SMZL: splenic marginal zone lymphoma; NNK: NF-κB/NOTCH/KLF2; SMZL: splenic marginal zone lymphoma; EPOCH: etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin; HIV: human immunodeficiency virus; EBV: Epstein-Barr virus; CD: cluster of differentiation; MUM1/IRF4: melanoma associated antigen (mutated) 1, interferon regulatory factor 4; MYC: myc proto-oncogene protein; BCL2: B-cell lymphoma 2; DLBCL: diffuse large B-cell lymphoma; DHL: double-hit lymphoma; AKI: acute kidney injury; CT: computed tomography; Hgb: hemoglobin; DAT: direct antiglobulin test; IgG: immunoglobulin G; LDH: lactate dehydrogenase; PET-CT: positron emission tomography-computed tomography; BM: bone marrow; SOX11: SRY-related HMG-box transcription factor 11; PAX5: paired box transcription factor 5; FISH: fluorescence in situ hybridization; IGH: immunoglobulin heavy chain; KLF2: Kruppel-like factor 2; CARD11: caspase recruitment domain-containing protein 11; MYD88: myeloid differentiation primary response 88; CXCR4: C-X-C chemokine receptor type 4; HHV-8: human gamma herpesvirus 8; ALK: anaplastic lymphoma kinase; BCL6: B-cell lymphoma 6; PCR: polymerase chain reaction; CMV: cytomegalovirus; HLH: hemophagocytic lymphohistiocytosis; MRI: magnetic resonance imaging; NGS: next-generation sequencing; FDG: 18F-fluorodeoxygllucose; ISH: in situ hybridization; R-IPI: revised international prognostic index; CNS-IPI: central nervous system international prognostic index; NCCN-IPI: National Comprehensive Cancer Network international prognostic index; CR: complete response; V-EPOCH: bortezomib, etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin; RTK: receptor tyrosine kinase; RAS: rat sarcoma virus; RAF: rapidly accelerated fibrosarcoma; JAK: janus kinase; STAT: signal transducer activator of transcription; MCL1: induced myeloid leukemia cell differentiation protein; IRF4: interferon regulatory factor 4; MAPK: mitogen-activated protein kinase; TP53: tumor protein 53; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; NTRK3: neurotrophic tyrosine kinase receptor type 3; POT1: protection of telomeres 1; VAF: variant allele frequencies
| References | ▴Top |
- Grimm KE, O'Malley DP. Aggressive B cell lymphomas in the 2017 revised WHO classification of tumors of hematopoietic and lymphoid tissues. Ann Diagn Pathol. 2019;38:6-10.
doi pubmed - Castillo JJ, Bibas M, Miranda RN. The biology and treatment of plasmablastic lymphoma. Blood. 2015;125(15):2323-2330.
doi pubmed - Chen BJ, Chuang SS. Lymphoid neoplasms with plasmablastic differentiation: a comprehensive review and diagnostic approaches. Adv Anat Pathol. 2020;27(2):61-74.
doi pubmed - Lopez A, Abrisqueta P. Plasmablastic lymphoma: current perspectives. Blood Lymphat Cancer. 2018;8:63-70.
doi pubmed - Ise M, Kageyama H, Ikebe D, Araki A, Kumagai K, Itami M. Transformation of double-hit follicular lymphoma to plasmablastic lymphoma: a partial role of MYC gene rearrangement. J Clin Exp Hematop. 2018;58(3):128-135.
doi pubmed - Martinez D, Valera A, Perez NS, Sua Villegas LF, Gonzalez-Farre B, Sole C, Gine E, et al. Plasmablastic transformation of low-grade B-cell lymphomas: report on 6 cases. Am J Surg Pathol. 2013;37(2):272-281.
doi pubmed - Li YJ, Li JW, Chen KL, Li J, Zhong MZ, Liu XL, Yi PY, et al. HIV-negative plasmablastic lymphoma: report of 8 cases and a comprehensive review of 394 published cases. Blood Res. 2020;55(1):49-56.
doi pubmed - Barouqa M, Greipp P, King R, McPhail ED. Plasmablastic lymphoma with MYC::IGH fusion and BCL2 rearrangement. EJHaem. 2022;3(3):1080-1081.
doi pubmed - Makady NF, Ramzy D, Ghaly R, Abdel-Malek RR, Shohdy KS. The emerging treatment options of plasmablastic lymphoma: analysis of 173 individual patient outcomes. Clin Lymphoma Myeloma Leuk. 2021;21(3):e255-e263.
doi pubmed - Debaugnies F, Mahadeb B, Ferster A, Meuleman N, Rozen L, Demulder A, Corazza F. Performances of the H-Score for diagnosis of hemophagocytic lymphohistiocytosis in adult and pediatric patients. Am J Clin Pathol. 2016;145(6):862-870.
doi pubmed - Bastidas-Mora G, Bea S, Navarro A, Gine E, Costa D, Delgado J, Baumann T, et al. Clinico-biological features and outcome of patients with splenic marginal zone lymphoma with histological transformation. Br J Haematol. 2022;196(1):146-155.
doi pubmed - Salido M, Baro C, Oscier D, Stamatopoulos K, Dierlamm J, Matutes E, Traverse-Glehen A, et al. Cytogenetic aberrations and their prognostic value in a series of 330 splenic marginal zone B-cell lymphomas: a multicenter study of the Splenic B-Cell Lymphoma Group. Blood. 2010;116(9):1479-1488.
doi pubmed - Bonfiglio F, Bruscaggin A, Guidetti F, Terzi di Bergamo L, Faderl M, Spina V, Condoluci A, et al. Genetic and phenotypic attributes of splenic marginal zone lymphoma. Blood. 2022;139(5):732-747.
doi pubmed - Clipson A, Wang M, de Leval L, Ashton-Key M, Wotherspoon A, Vassiliou G, Bolli N, et al. KLF2 mutation is the most frequent somatic change in splenic marginal zone lymphoma and identifies a subset with distinct genotype. Leukemia. 2015;29(5):1177-1185.
doi pubmed - Pich A, Fraire F, Fornari A, Bonino LD, Godio L, Bortolin P, Chiusa L, et al. Intrasinusoidal bone marrow infiltration and splenic marginal zone lymphoma: a quantitative study. Eur J Haematol. 2006;76(5):392-398.
doi pubmed - Vela V, Juskevicius D, Dirnhofer S, Menter T, Tzankov A. Mutational landscape of marginal zone B-cell lymphomas of various origin: organotypic alterations and diagnostic potential for assignment of organ origin. Virchows Arch. 2022;480(2):403-413.
doi pubmed - Morner-Svalling AC, Tronje G, Andersson LG, Welander U. Comparison of the diagnostic potential of direct digital and conventional intraoral radiography in the evaluation of peri-implant conditions. Clin Oral Implants Res. 2003;14(6):714-719.
doi pubmed - Kim H, Kim HJ, Kim SH. Diagnostic approach for double-hit and triple-hit lymphoma based on immunophenotypic and cytogenetic characteristics of bone marrow specimens. Ann Lab Med. 2020;40(5):361-369.
doi pubmed - Castillo JJ, Guerrero-Garcia T, Baldini F, Tchernonog E, Cartron G, Ninkovic S, Cwynarski K, et al. Bortezomib plus EPOCH is effective as frontline treatment in patients with plasmablastic lymphoma. Br J Haematol. 2019;184(4):679-682.
doi pubmed - Bartlett NL, Wilson WH, Jung SH, Hsi ED, Maurer MJ, Pederson LD, Polley MC, et al. Dose-adjusted EPOCH-R compared with R-CHOP as frontline therapy for diffuse large B-cell lymphoma: clinical outcomes of the phase III intergroup trial alliance/CALGB 50303. J Clin Oncol. 2019;37(21):1790-1799.
doi pubmed - Frontzek F, Staiger AM, Zapukhlyak M, Xu W, Bonzheim I, Borgmann V, Sander P, et al. Molecular and functional profiling identifies therapeutically targetable vulnerabilities in plasmablastic lymphoma. Nat Commun. 2021;12(1):5183.
doi pubmed - Ramis-Zaldivar JE, Gonzalez-Farre B, Nicolae A, Pack S, Clot G, Nadeu F, Mottok A, et al. MAPK and JAK-STAT pathways dysregulation in plasmablastic lymphoma. Haematologica. 2021;106(10):2682-2693.
doi pubmed - Witte HM, Kunstner A, Hertel N, Bernd HW, Bernard V, Stolting S, Merz H, et al. Integrative genomic and transcriptomic analysis in plasmablastic lymphoma identifies disruption of key regulatory pathways. Blood Adv. 2022;6(2):637-651.
doi pubmed - Weeks AC, Kimple ME. Spontaneous Tumor Lysis Syndrome: A Case Report and Critical Evaluation of Current Diagnostic Criteria and Optimal Treatment Regimens. J Investig Med High Impact Case Rep. 2015;3(3):2324709615603199.
doi pubmed - Chapman J, Gentles AJ, Sujoy V, Vega F, Dumur CI, Blevins TL, Bernal-Mizrachi L, et al. Gene expression analysis of plasmablastic lymphoma identifies downregulation of B-cell receptor signaling and additional unique transcriptional programs. Leukemia. 2015;29(11):2270-2273.
doi pubmed - Hertel N, Merz H, Bernd HW, Bernard V, Kunstner A, Busch H, von Bubnoff N, et al. Performance of international prognostic indices in plasmablastic lymphoma: a comparative evaluation. J Cancer Res Clin Oncol. 2021;147(10):3043-3050.
doi pubmed - Yanamandra U, Sahu KK, Jain N, Prakash G, Saikia U, Malhotra P. Plasmablastic lymphoma: successful management with CHOP and lenalidomide in resource constraint settings. Ann Hematol. 2016;95(10):1715-1717.
doi pubmed - Ryu YK, Ricker EC, Soderquist CR, Francescone MA, Lipsky AH, Amengual JE. Targeting CD38 with daratumumab plus chemotherapy for patients with advanced-stage plasmablastoid large B-cell lymphoma. J Clin Med. 2022;11(16):4928.
doi pubmed - Castillo JJ, Lamacchia J, Silver J, Flynn CA, Sarosiek S. Complete response to pembrolizumab and radiation in a patient with HIV-negative, EBV-positive plasmablastic lymphoma. Am J Hematol. 2021;96(10):E390-E392.
doi
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.