

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://www.thejh.org |
Case Report
Volume 12, Number 2, April 2023, pages 82-86

De-Novo B-Cell Prolymphocytic Leukemia
Sasmith R. Menakurua, d, Janet Roepkeb, Salahuddin Siddiquic
aDepartment of Internal Medicine, Indiana University School of Medicine-Muncie, Muncie, IN, USA
bDepartment of Pathology, Indiana University Health-Ball Memorial Hospital, Muncie, IN, USA
cDepartment of Hematology/Oncology, Indiana University Health-Ball Memorial Hospital, Muncie, IN, USA
dCorresponding Author: Sasmith R. Menakuru, Department of Internal Medicine, Indiana University School of Medicine-Muncie, Muncie, IN, USA
Manuscript submitted January 27, 2023, accepted April 5, 2023, published online April 30, 2023
Short title: De-Novo B-PLL
doi: https://doi.org/10.14740/jh1096
| Abstract | ▴Top |
B-cell prolymphocytic leukemia (B-PLL) is a rare B-cell neoplasm that typically presents with splenomegaly, a rising white blood cell count, and may or may not have B symptoms. The diagnosis usually requires a bone marrow biopsy and aspirate with flow cytometry and cytogenetic studies. At least 55% of the lymphocytes in the peripheral blood must be prolymphocytes to be defined as B-PLL. A thorough differential diagnosis would include mantle cell lymphoma, chronic lymphocytic leukemia (CLL) with prolymphocytes, hairy cell leukemia, and splenic marginal zone lymphoma. B-PLL is managed with regimens utilized for CLL, such as ibrutinib and rituximab but is tailored for each individual. The authors report a rare case of B-PLL in a patient with no known history of CLL. The authors discuss this entity in context of the 2017 and 2022 World Health Organization (WHO) classifications, the latter of which no longer recognizes B-PLL as a distinct entity. The authors hope that this article helps practitioners with the diagnosis and treatment of B-PLL. Perhaps with better recognition, and better documentation of histopathologic features of these rare cases going forward, it may prove to be a distinct entity again in future classifications.
Keywords: B-cell prolymphocytic leukemia; Differential diagnosis; Treatment
| Introduction | ▴Top |
B-cell prolymphocytic leukemia (B-PLL) accounts for less than 1% of all lymphoid leukemias, making it extremely rare in clinical practice, with only around 120 cases in the United States yearly [1]. As will be discussed later, the new 2022 World Health Organization (WHO) classifications no longer recognizes this entity and has put it along with hairy cell leukemia variant (HCL-v) into a new category called splenic B-cell lymphoma with prominent nucleoli (SBLPN). The number of cases in this new category is low and accounts for approximately 0.4% of chronic lymphoid malignancies [2-5]. And most of those cases had been previously classified as HCL-v. Therefore, the number of cases of B-PLL (that would now belong in the SBLPN category) would account for even less than 0.4% of chronic lymphoid malignancies by our estimation.
B-cell PLL (prior to the 2022 WHO classification) was typically characterized as very aggressive, having TP53 dysfunction, affecting older adults with a median age of 69, and is slightly more common in men. B-PLL may have similarities to chronic lymphocytic leukemia (CLL) and mantle cell lymphoma (MCL) in the leukemic phase but can be distinguished from each other based on immunophenotypic and cytogenetic results. The characteristic of B-PLL is the presence of prolymphocyte morphology of greater than 55% of the lymphocytes in the blood or bone marrow [6]. B-PLL will also have a brighter CD20 expression compared to CLL’s dim CD20 expression. Prolymphocytes in the peripheral blood are commonly seen in patients with CLL, but in those cases, the percentage of lymphocytes with prolymphocyte morphology is usually low and definitely less than 55%. In most instances, B-cell PLL occurs as a transformation or evolution of a slow-growing CLL and is extremely rare as a primary disorder, which we believe to have occurred in this case study example. The prognosis of B-PLL is generally poor, with a median survival of 3 years [7]. The authors describe a case of an aggressive de-novo B-PLL with splenomegaly.
| Case Report | ▴Top |
A 71-year-old male was referred to hematology by his primary care provider for a complete blood count (CBC) showing a platelet count of 64,000/µL, a white blood cell (WBC) count of 48,700/µL, and an absolute monocyte count of 36,000/µL. His laboratory investigations previously did not show any abnormalities with his CBC within normal limits 3 months prior. He also said that he had lost 25 pounds in the past 2 months. He denied fevers, chills, night sweats, swollen glands, or appetite changes. An abdominal computed tomography (CT) showed a significantly enlarged spleen of 19 cm. The patient’s coagulation profile, liver and renal function, viral testing for hepatitis and human immunodeficiency virus (HIV), and autoimmune profile were unrevealing.
The bone marrow biopsy from the right iliac crest showed extensive involvement by atypical lymphocytes with round nuclear contours, prominent nucleoli, and moderate to abundant cytoplasm. Immunohistochemical stain for PAX5 demonstrated that the atypical lymphocytes accounted for 60% of the total cellularity. Additional immunohistochemical stains demonstrated that the atypical lymphocytes were positive for BCL2 and negative for BCL6, LMO2, and cyclin D1. The proliferation index by Ki67 was 5-10%. The peripheral blood for flow cytometry showed a prominent monoclonal B-cell population with a high-side light scatter. The B cells were positive for CD19, CD20, CD22, human leukocyte antigen-DR (HLA-DR), and bright kappa light chains. They were negative for CD10, CD5, CD23, CD25, CD11C, CD103, and CD123. Fluorescence in situ hybridization (FISH) showed both the loss of 17p13.1 (p53) and 13q14. Loss of 13q14 is not specific as it can be seen in other lymphomas such as CLL, but it is also seen in 27% of cases of B-PLL. The diagnosis of B-PLL is often a challenge due to an overlap with other mature B-cell leukemias and lymphomas, and therefore a broad differential diagnosis is needed (Fig. 1). The morphological features, immunophenotypic features, and clinical findings of splenomegaly were consistent with de-novo B-PLL (Figs. 2-4).
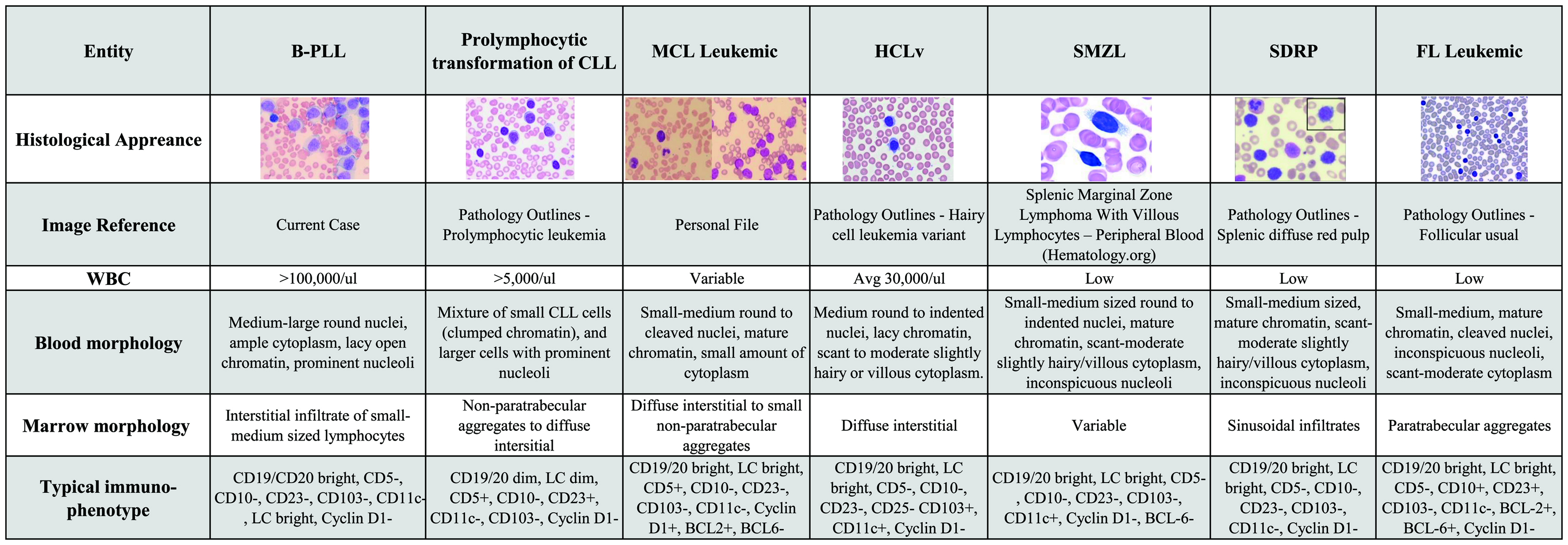 Click for large image | Figure 1. A broad differential diagnosis for B-PLL. B-PLL: B-cell prolymphocytic leukemia; CLL: chronic lymphocytic leukemia; MCL: mantle cell lymphoma; HCL-v: hairy cell leukemia variant; SMZL: splenic marginal zone lymphoma; SDRP: splenic diffuse red pulp; FL: follicular lymphoma; WBC: white blood cell; Avg: average; LC: light chain. |
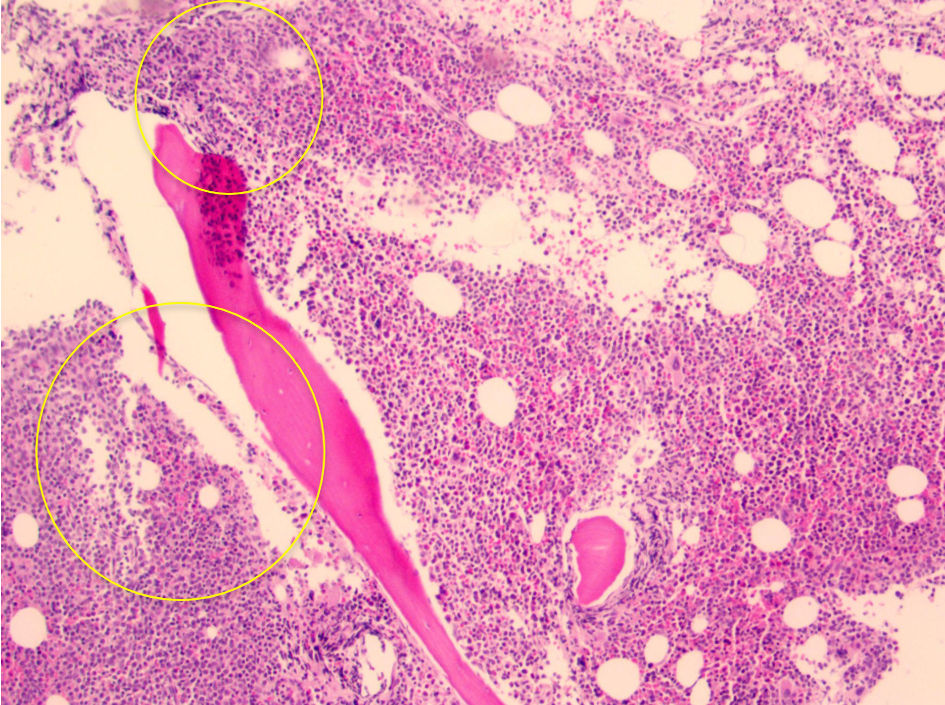 Click for large image | Figure 2. Core biopsy showing hypercellular marrow for age of patient (about 90%), with ill-defined aggregates of lymphocytes (yellow circled areas). Note the lack of “fried-egg” appearance which is seen with hairy cell leukemia. |
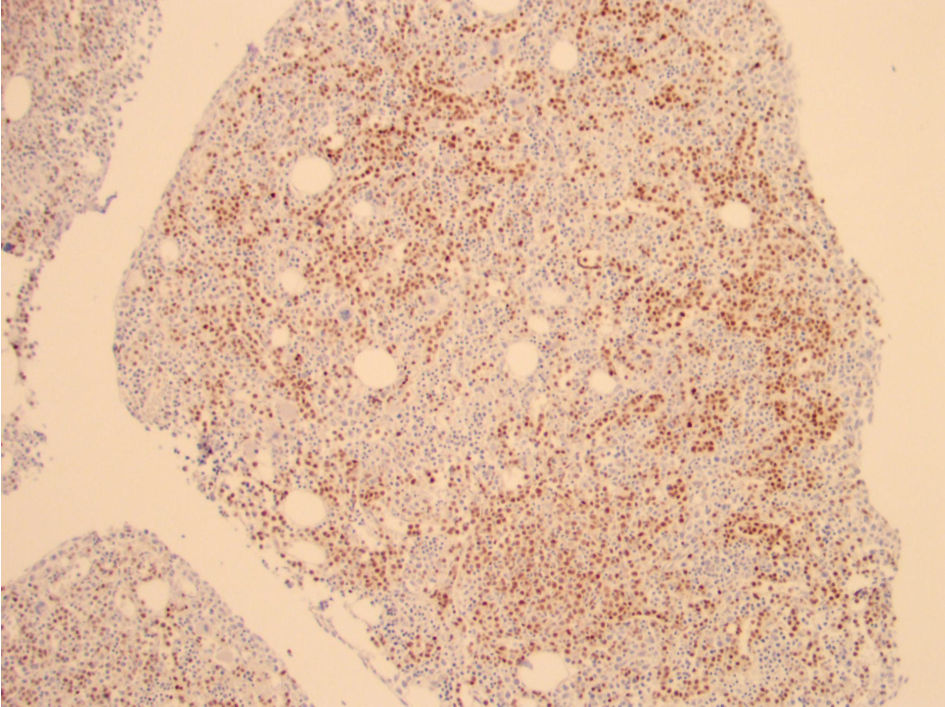 Click for large image | Figure 3. Pax 5 immunohistochemical stain showing increased B cells (brown staining). Note the lack of intrasinusoidal distribution of B cells typically seen in splenic diffuse red pulp lymphoma. |
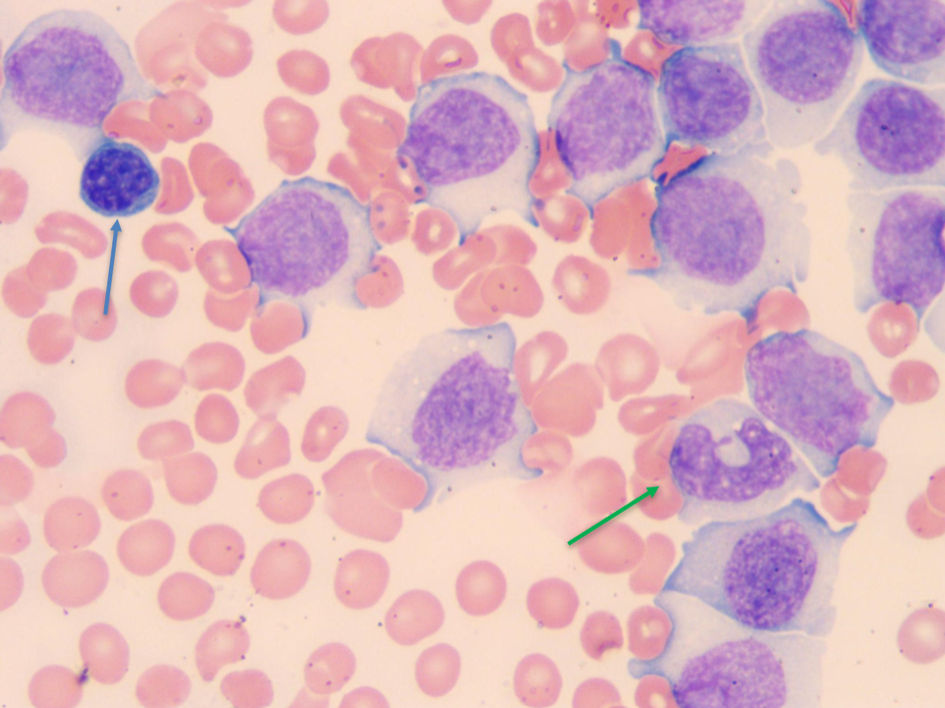 Click for large image | Figure 4. Blood smear showing a normal small lymphocyte near the upper left-hand corner (blue arrow), and normal monocytes near the lower right corner (green arrow) for comparison. The rest of the cells are B-PLL cells which are as big if not bigger than monocytes. B-PLL: B-cell prolymphocytic leukemia. |
He was started on ibrutinib 420 mg daily due to his 17p loss and allopurinol 300 mg twice daily due to an elevated uric acid. The patient’s thrombocytopenia at the time of diagnosis could be due to splenic sequestration, immune suppression, or immune destruction, as his bone marrow showed adequate megakaryopoiesis. His thrombocytopenia worsened, and his WBC counts increased; therefore, rituximab was added to his regimen. His ibrutinib was held due to his thrombocytopenia. His thrombocytopenia did not improve despite multiple transfusions, and it was deemed refractory. Unfortunately, given his rapid clinical deterioration within 2 weeks of diagnosis, conventional karyotyping, immunoglobulin heavy chain gene mutations, and molecular abnormality testing by next-generation sequencing could not be performed. The patient did not show significant improvement and deteriorated clinically. He elected to go comfort care as he did not want to try any other treatment options and passed away 4 months after his diagnosis.
| Discussion | ▴Top |
First described in 1974 by Galton et al, B-PLL is a neoplasm of B cells comprised of prolymphocytes, typically involving the bone marrow, spleen, and peripheral blood, which the WHO recognized in the previous (2017, fourth) edition of the classification of hematolymphoid diseases [8]. It is a clinically aggressive neoplasm with a poor prognosis that occurs in older patients with B-type symptoms and typically has splenomegaly with minimal to absent peripheral lymphadenopathy. Despite being called prolymphocytic, which suggests the cells might be immature, the cells involved in this condition are mature activated B cells that are large cells with mature, moderately dense chromatin, a large prominent vesicular nucleolus, and usually abundant non-villous, non-hairy cytoplasm [9].
The diagnosis of B-PLL is usually made based on the results of immunophenotypic and genetic analysis of peripheral blood, and by definition, at least 55% of the lymphocytes in the blood should have prolymphocyte morphology. Results of a bone marrow biopsy can confirm the findings, and a splenectomy can be diagnostic in patients with an unclear presentation with an enlarged spleen. Lymph node biopsies, in most cases, will not show any additional information. Utilizing flow cytometry, the cells will demonstrate a nonspecific B-cell immunophenotype with light chain restriction, bright surface immunoglobulin, and the expression of B-cell antigens. CD5 and CD23 are usually absent but can be weakly expressed; however, CD11c, CD103, CD10, and CD25 are not expressed.
The differential diagnosis based on morphology would also include T-cell PLL, MCL with prolymphocytoid transformation, splenic marginal zone lymphoma (SMZL), CLL with prolymphocytoid transformation, CD10-negative follicular lymphoma (FL), hairy cell leukemia (HCL), and HCL-v. T-PLL is easily distinguished as a well-defined clinical entity as it expresses T-cell antigens in contrast to B [10]. With the 2017 WHO classification, the number of cases meeting criteria for B-PLL decreased further as many would be reclassified as HCL-v, splenic diffuse red pulp lymphoma (SDRPL), MCL with prolymphocyte morphology, or atypical CLL [10, 11]. CLL/PLL was previously defined as CLL with between 15% and 55% of prolymphocytes but has been removed from the more recent 2017 and 2022 WHO classifications of lymphoid neoplasms. CLL was the first B-cell lymphoma that was recognized to undergo prolymphocytic transformation, and the pathological features were indistinguishable from de-novo B-PLL. These cases in the 2017 and 2022 WHO classifications are referred to as CLL with increased lymphocytes with prolymphocytic morphology. In such cases, the peripheral blood contains a mixture of small mature CLL cells and prolymphocytes. These prolymphocytes will have an immunophenotype that is similar to CLL. In CLL, the prolymphocytes are seen in variable numbers but must be 55% or less of the circulating cells, and as a rule, the prolymphocytes of CLL show histological and immunophenotypic findings consistent with CLL, such as having pseudo-proliferation centers on biopsy specimens and expressing CD5 and CD23 [12]. In contrast, B-PLL cells are monomorphic large lymphocytes with an immunophenotype that is not characteristic of CLL, MCL, FL, or HCL.
MCL may have a leukemic phase; however, the neoplastic cells of MCL will express CD5, and cyclin D1, the latter of which is due to (11;14) translocation of the cyclin D1 gene to the IgH gene [6]. In contrast, B-PLL’s malignant cells are negative for CD5, cyclin D1, and do not demonstrate (11;14) translocation. Rare cases of t(11;14) (q13;q32) MCL with prolymphocytic features have been called a prolymphocytic variant of MCL.
SMZL and B-PLL can present similarly with splenomegaly and peripheral blood lymphocytosis. SMZL is less aggressive, and bone marrow morphology in SMZL may take the form of reactive appearing follicles surrounded by marginal zone B cells, which helps differentiate it from B-PLL [13]. SMZL cells in the peripheral blood are smaller than in B-PLL cells, do not have prominent nucleoli, and often have villous appearing cytoplasmic borders. In difficult cases where the differentiation is unclear, mutational testing for genes such as NOTCH2 may be helpful, as well as bone marrow, spleen, and hilar lymph node evaluation. NOTCH2 is mutated in 10-25% of cases of MZL.
HCL has similarities to B-PLL, including lymphocytes that are present in the monocyte gate in flow cytometry, and patients typically have splenomegaly and peripheral blood cytopenias. HCL patients can have lymphocytosis but often have relatively normal or low peripheral blood lymphocyte counts. HCL is easily distinguished from B-PLL by flow cytometry as HCL expresses CD25, CD103, CD11c, and demonstrates positive immunohistochemical staining for DBA44, annexin A1, and tartrate resistant acid phosphatase (TRAP).
HCL-v is difficult to distinguish from B-PLL, and in fact, the 2022 fifth edition of the WHO classification combines B-PLL and HCL-v into a new category called splenic B-cell leukemia/lymphoma with prominent nucleoli (SBLPN). Therefore, in the new WHO classification, this case report would be classified as SBLPN [14]. The distinction may not be relevant clinically at this time, which is one argument for combining these similar entities in the 2022 WHO classification. It will be interesting to see over time and with more case reports of SBLPN if diagnostic features can be identified that can distinguish B-PLL from HCL-v. In the 2017 WHO classification, HCL-v was believed to express CD11c and CD103, which would distinguish it from B-PLL, which does not express these antigens [11].
A CD10-negative FL in leukemic phase is not likely to be easily confused with B-PLL as it typically has dense chromatin, cleaved nuclei, and a higher nucleus-to-cytoplasmic ratio. FL that has transformed into large-cell lymphoma with immunoblastic morphology, and is in the leukemic phase could be confused morphologically with B-PLL. However, FL is usually less monomorphic, expresses BCL-6 even if it does not express CD10, usually demonstrates t(14;18) translocation, and not abnormalities of 13q14 as in this case.
Treatment for B-PLL is variable depending on each individual case, as some patients may be asymptomatic without complications, treatment may be deferred initially; however, treatment is indicated in patients with complications such as a rapidly rising WBC count, splenomegaly, and B symptoms. Given the rarity of B-PLL, there are no current guidelines on treatment, and therefore, most commonly, it is treated similarly to regimens utilized for CLL [15]. Responses to treatment are highly varied, and unfortunately, most frequently they are partial and rarely prolonged. The choice of treatment is dependent on the individual and the presence of high-risk genetic features such as deletion of 17p or TP53 mutations [16].
Retrospective reviews have suggested that B-PLL is a highly variable disorder, with some cases having an aggressive course and others being more indolent [17]. This may be in part due to cases diagnosed as B-PLL representing more than one type of lymphoma. For patients without deletion of 17p or TP53 mutations, a possible regimen may be utilizing fludarabine, cyclophosphamide, rituximab (FCR), or bendamustine, rituximab (BR). In older individuals and those with comorbidities, ibrutinib may be considered. Rituximab can be utilized in individuals who cannot receive chemotherapy and may provide palliation [18]. In patients with deletion of 17p or TP53 mutations, ibrutinib is the choice of treatment; however, alemtuzumab may be considered as well [19]. In this case, ibrutinib was chosen because the patient was elderly and had a 17p deletion. Rituximab was added given the lack of response to ibrutinib and the overall worsening of his clinical status.
Hematopoietic stem cell transplantation can be considered in young, fit patients who have responded to initial therapy and those with poor responses to initial therapy [20]. Refractory and/or replaced disease can be possibly managed with ibrutinib, idelalisib, and venetoclax [21]. Currently, the data gathered from B-PLL treatment are limited to case reports and case series with few participants. In a case series, five patients were treated with idelalisib combined with rituximab, and all five patients had a response lasting greater than 6 months [22]. A case report by Xing et al treated a patient with B-PLL with zanubrutinib, rituximab, and lenalidomide, which provided a durable response [23]. A case report by Siddiqui et al treated a patient with ibrutinib and venetoclax, which provided a positive response for 3 years [24]. Hew et al utilized obinutuzumab-chlorambucil which showed a sustained response as well [25]. Cases of SBLPN have shown responses to cladribine/rituximab or cladribine/bendustamine combinations [26]. Currently, clinical trials are in progress to evaluate the role of chimeric antigen receptor-modified T cells (CAR-T).
The authors hope this case report and review of the current limited literature provide physicians with diagnostic tools and possible treatment options for B-PLL. Optimal treatment for B-PLL requires large-scale clinical trials, but the rarity of the disease limits this. We hope that new research in future will provide accurate guidelines for treatment regimens. And again, careful classification and documentation of clinicopathologic features in cases of SBLPN may help determine if this category should remain as one or if subcategories such as HCL-v and B-PLL should be recognized.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
No conflict of interest to declare.
Informed Consent
Informed consent has been obtained from the patient.
Author Contributions
SRM saw the case, did research on the topic, and wrote the paper. JP and SS supervised and edited the paper.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Dearden C. How I treat prolymphocytic leukemia. Blood. 2012;120(3):538-551.
doi pubmed - Matutes E, Wotherspoon A, Catovsky D. The variant form of hairy-cell leukemia. Best Pr Res Clin Haematol. 2003;16:41-56.
- Tran J, Gaulin C, Tallman MS. Advances in the treatment of hairy cell leukemia variant. Curr Treat Options Oncol. 2022;23(1):99-116.
doi pubmed - Matutes E, Wotherspoon A, Brito-Babapulle V, Catovsky D. The natural history and clinico-pathological features of the variant form of hairy cell leukemia. Leukemia. 2001;15(1):184-186.
doi pubmed - Ludescher C, Gattringer C, Weger AR, Drach J, Thaler J, Bitschmann R, Huber H. Surface immunoglobulin density in the differential diagnosis of B-cell chronic lymphocytic leukemia and leukemic immunocytoma. Leuk Res. 1992;16(2):191-196.
doi pubmed - van der Velden VH, Hoogeveen PG, de Ridder D, Schindler-van der Struijk M, van Zelm MC, Sanders M, Karsch D, et al. B-cell prolymphocytic leukemia: a specific subgroup of mantle cell lymphoma. Blood. 2014;124(3):412-419.
doi pubmed - Hercher C, Robain M, Davi F, Garand R, Flandrin G, Valensi F, Vandeputte H, et al. A multicentric study of 41 cases of B-prolymphocytic leukemia: two evolutive forms. Leuk Lymphoma. 2001;42(5):981-987.
doi pubmed - Galton DA, Goldman JM, Wiltshaw E, Catovsky D, Henry K, Goldenberg GJ. Prolymphocytic leukaemia. Br J Haematol. 1974;27(1):7-23.
doi pubmed - Oscier D, Else M, Matutes E, Morilla R, Strefford JC, Catovsky D. The morphology of CLL revisited: the clinical significance of prolymphocytes and correlations with prognostic/molecular markers in the LRF CLL4 trial. Br J Haematol. 2016;174(5):767-775.
doi pubmed pmc - El Hussein S, Khoury JD, Medeiros LJ. B-prolymphocytic leukemia: is it time to retire this entity? Ann Diagn Pathol. 2021;54:151790.
doi pubmed - Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues, revised 4th edition. International Agency for Research on Cancer (IARC), Lyon. 2017.
- Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, Sultan C. Proposals for the classification of chronic (mature) B and T lymphoid leukaemias. French-American-British (FAB) Cooperative Group. J Clin Pathol. 1989;42(6):567-584.
doi pubmed pmc - Hoehn D, Miranda RN, Kanagal-Shamanna R, Lin P, Medeiros LJ. Splenic B-cell lymphomas with more than 55% prolymphocytes in blood: evidence for prolymphocytoid transformation. Hum Pathol. 2012;43(11):1828-1838.
doi pubmed - Alaggio R, Amador C, Anagnostopoulos I, Attygalle AD, Araujo IBO, Berti E, Bhagat G, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia. 2022;36(7):1720-1748.
doi pubmed pmc - van den Brand M, van Krieken JH. Recognizing nodal marginal zone lymphoma: recent advances and pitfalls. A systematic review. Haematologica. 2013;98(7):1003-1013.
doi pubmed pmc - Ravandi F, O'Brien S. Chronic lymphoid leukemias other than chronic lymphocytic leukemia: diagnosis and treatment. Mayo Clin Proc. 2005;80(12):1660-1674.
doi pubmed - Shvidel L, Shtalrid M, Bassous L, Klepfish A, Vorst E, Berrebi A. B-cell prolymphocytic leukemia: a survey of 35 patients emphasizing heterogeneity, prognostic factors and evidence for a group with an indolent course. Leuk Lymphoma. 1999;33(1-2):169-179.
doi pubmed - Mourad YA, Taher A, Chehal A, Shamseddine A. Successful treatment of B-cell prolymphocytic leukemia with monoclonal anti-CD20 antibody. Ann Hematol. 2004;83(5):319-321.
doi pubmed - Chaar BT, Petruska PJ. Complete response to alemtuzumab in a patient with B prolymphocytic leukemia. Am J Hematol. 2007;82(5):417.
doi pubmed - Castagna L, Sarina B, Todisco E, Mazza R, Santoro A. Allogeneic peripheral stem-cell transplantation with reduced-intensity conditioning regimen in refractory primary B-cell prolymphocytic leukemia: a long-term follow-up. Bone Marrow Transplant. 2005;35(12):1225.
doi pubmed - Damlaj M, Al Balwi M, Al Mugairi AM. Ibrutinib therapy is effective in B-cell prolymphocytic leukemia exhibiting MYC aberrations. Leuk Lymphoma. 2018;59(3):739-742.
doi pubmed - Eyre TA, Fox CP, Boden A, Bloor A, Dungawalla M, Shankara P, Went R, et al. Idelalisib-rituximab induces durable remissions in TP53 disrupted B-PLL but results in significant toxicity: updated results of the UK-wide compassionate use programme. Br J Haematol. 2019;184(4):667-671.
doi pubmed - Xing L, He Q, Xie L, Wang H, Li Z. Zanubrutinib, rituximab and lenalidomide induces deep and durable remission in TP53-mutated B-cell prolymphocytic leukemia: a case report and literature review. Haematologica. 2022;107(5):1226-1228.
doi pubmed pmc - Siddiqui MT, Price A, Ferrajoli A, Borthakur G. Sustained MRD negative remission in del17p and TP53 mutated B cell prolymphocytic leukemia with ibrutinib and venetoclax. Leuk Res Rep. 2021;16:100266.
doi pubmed pmc - Hew J, Pham D, Matthews Hew T, Minocha V. A novel treatment with Obinutuzumab-chlorambucil in a patient with B-cell prolymphocytic leukemia: a case report and review of the literature. J Investig Med High Impact Case Rep. 2018;6:2324709618788674.
doi pubmed pmc - Maitre E, Paillassa J, Troussard X. Novel targeted treatments in hairy cell leukemia and other hairy cell-like disorders. Front Oncol. 2022;12:1068981.
doi pubmed pmc
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.