

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://www.thejh.org |
Case Report
Volume 12, Number 5, October 2023, pages 236-242

When Lymphoma Strikes the Pancreas: A Rare Presentation of Systemic Anaplastic Lymphoma Kinase-Negative Anaplastic Large Cell Lymphoma in a Human Immunodeficiency Virus-Positive Patient
Hehua Hannah Huanga, b, Xin Qinga, b ![]()
aDepartment of Pathology and Laboratory Medicine, Harbor-UCLA Medical Center, Torrance, CA 90502, USA
bCorresponding Author: Hehua Huang and Xin Qing, Department of Pathology and Laboratory Medicine, Harbor-UCLA Medical Center, Torrance, CA 90502, USAand
Manuscript submitted May 31, 2023, accepted September 4, 2023, published online October 21, 2023
Short title: Rare ALK-Negative ALCL in HIV-Positive Patient
doi: https://doi.org/10.14740/jh1138
| Abstract | ▴Top |
Anaplastic lymphoma kinase (ALK)-negative anaplastic large cell lymphoma (ALCL) is an uncommon subtype of non-Hodgkin lymphoma, with pancreatic involvement being exceedingly rare and documented in only a handful of case reports. We present a unique case of a 31-year-old human immunodeficiency virus (HIV)-positive male with multisite ALK-negative ALCL, who initially presented with a buttock ulcer, leading to a suspicion of primary cutaneous ALCL or lymphomatoid papulosis. However, the discovery of multiple extracutaneous sites, including an atypical pancreatic head involvement, confirmed the diagnosis of systemic ALK-negative ALCL with cutaneous manifestation. The patient received six cycles of brentuximab vedotin + cyclophosphamide-doxorubicin-prednisone (BV + CHP) treatment, achieving a substantial reduction in the size of the pancreatic head mass and no detectable fluorodeoxyglucose (FDG) uptake on positron emission tomography (PET) scan. This case underscores the diagnostic challenges of ALK-negative ALCL in HIV-positive patients with extranodal presentations and demonstrates the potential effectiveness of targeted therapeutic strategies for such cases.
Keywords: ALK-negative anaplastic large cell lymphoma; Systemic ALK-negative ALCL; HIV; Non-Hodgkin lymphoma; Pancreatic involvement; Cutaneous manifestation
| Introduction | ▴Top |
Anaplastic large cell lymphoma (ALCL) represents a rare type of mature T-cell lymphomas, hallmarked by pleomorphic tumor cells with uniform strong expression of CD30 antigen [1]. According to the most recent World Health Organization (WHO) classification from 2022, there are three entities within the family of ALCL: anaplastic lymphoma kinase (ALK)-positive ALCL, ALK-negative ALCL, and breast implant-associated ALCL [2]. Primary cutaneous ALCL is grouped under primary cutaneous T-cell lymphoma [2]. ALK-negative ALCL constitutes approximately 30% of all ALCL cases [1]. Compared to ALK-positive ALCL, ALK-negative ALCL is less prevalent and exhibits a poorer prognosis [3, 4].
Systemic ALK-negative ALCL is typified by the absence of ALK gene rearrangements, often manifesting systemic symptoms such as fever, night sweats, weight loss, lymphadenopathy, and extranodal involvement [1]. Its prognosis is less favorable than ALK-positive ALCL, with a 5-year survival rate ranging from 40% to 60% [3].
Involvement of the pancreas, particularly the pancreatic head, is an exceedingly rare event in ALCL. Primary pancreatic lymphoma accounts for less than 0.5% of all pancreatic neoplasms, with only a handful of reported ALK-negative ALCL cases involving the pancreas in the literature [5, 6].
Diagnosing ALK-negative ALCL poses a challenge due to its diverse clinical and histopathological presentations, which can complicate its differentiation from other T-cell lymphomas and benign or reactive conditions [7]. We describe a case of systemic ALK-negative ALCL involving a 31-year-old human immunodeficiency virus (HIV)-positive male, who initially presented with a skin ulcer but subsequently was found to have pancreatic head involvement. The diagnosis of ALK-negative ALCL involving the pancreas presents challenges not only for dermatopathologists and gastrointestinal (GI) specialists but also for hematopathologists, owing to the rarity of this manifestation and the morphological overlap of large atypical cells with other neoplasms [7].
| Case Report | ▴Top |
A 31-year-old male presented with a 2-year history of recurrent perirectal ulceration. His recent health deteriorated over the span of 3 weeks, marked by generalized sluggishness and diarrhea, culminating in an acute altered mental status (AMS) lasting 24 h that prompted his family to seek urgent medical intervention.
Upon admission to the intensive care unit (ICU) due to shock and acute encephalopathy, a fresh diagnosis of HIV was established. Diagnostic workup revealed a critically low CD4+ count of 45 cells/mm3 and a high viral load exceeding 2 million copies/mL of genotype B/wild type. While the patient developed oral thrush during hospitalization, he displayed no other opportunistic infections. Accordingly, the medical team initiated antiretroviral therapy (ART) with TAF/FTC + DTG (dolutegravir and emtricitabine-tenofovir).
Blood investigations further painted a concerning picture with an absolute lymphocyte count below 1,000/mm3. Radiographic studies, including computed tomography (CT) scans, revealed hepatomegaly and multiple mediastinal and axillary lymph nodes of varied dimensions. The triad of fever, shock, and AMS initially oriented the medical team towards a potential infectious etiology. However, extensive diagnostic evaluations ruled out infectious causes, and the patient’s condition stabilized with supportive care.
The patient’s neurological manifestations, characterized by profound amnesia, a state of withdrawal, and bilateral hippocampal changes on imaging, aligned with a diagnosis of limbic/autoimmune encephalitis. Therapeutic plasmapheresis was employed across five sessions, leading to significant clinical improvement, although the patient had not yet fully returned to his baseline health.
Retrospective assessment postulated that his clinical manifestations could have been triggered by an immune response to an underlying malignancy. This hypothesis was formulated even though specific antibodies such as anti-Hu, Ri, Yo, anti-N-methyl-D-aspartate receptor (NMDAR) and anti-glutamic acid decarboxylase (anti-GAD 65) were undetected.
Upon skin examination, persistent ulcers were noted on the left medial buttock. These ulcers presented with beefy-red granulation tissue, defined edges, minimal exudate, and lacked tenderness or discernible induration. Despite receiving antibiotic and antiviral therapies since admission, ulcerations on the right buttock remained unchanged. Tests for cytomegalovirus (CMV), herpes simplex virus (HSV) polymerase chain reaction (PCR), and fungal serologies came back negative.
Histopathological assessment of the ulcer biopsy highlighted sporadically grouped large, atypical cells interspersed with inflammatory cells like lymphocytes and histiocytes. Importantly, the so-called “hallmark” cells, characterized by their distinctively large pleomorphic structure and a unique horseshoe-shaped nucleus indicative of ALCL, were subtle in the skin biopsy (Fig. 1). Benharroch et al first described these cells in 1998 [8].
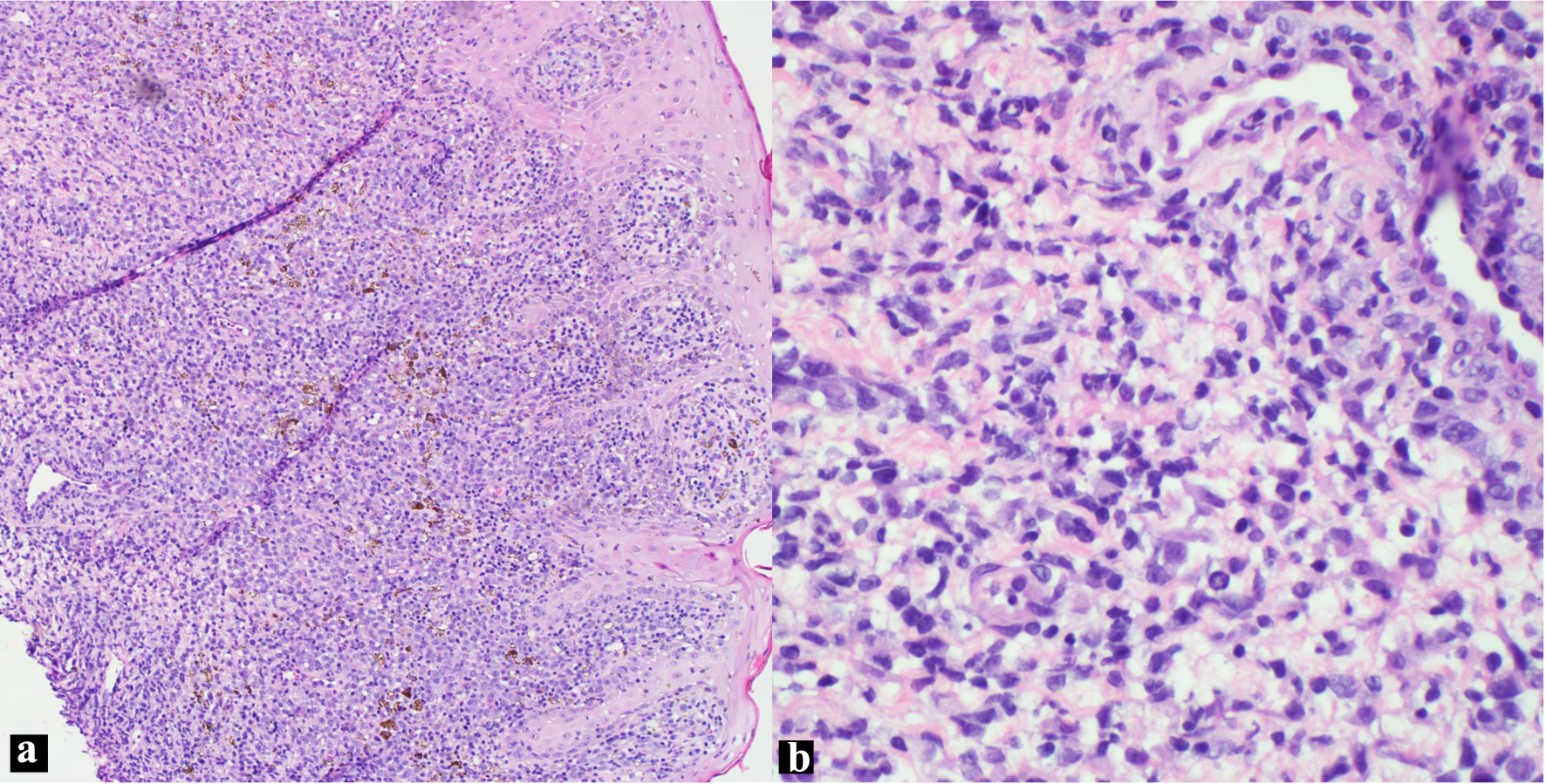 Click for large image | Figure 1. H&E skin biopsy (a: × 100; b: × 400 magnification), showing non-cohesive, anaplastic pleomorphic cells. H&E: hematoxylin and eosin. |
Immunohistochemistry (IHC) staining (Fig. 2) revealed these large cells tested positive for CD30, CD2, CD4, CD5, CD43, and MUM-1 markers, while they were negative for ALK-1, CD3, CD8, EMA, CD20, and PAX-5. Their status for CD45 was inconclusive. The primary differential diagnoses considered included primary cutaneous ALCL, lymphomatoid papulosis (LyP), and systemic ALK-negative ALCL with skin involvement. A bone marrow biopsy, conducted to assess pancytopenia, showed no signs of lymphoma.
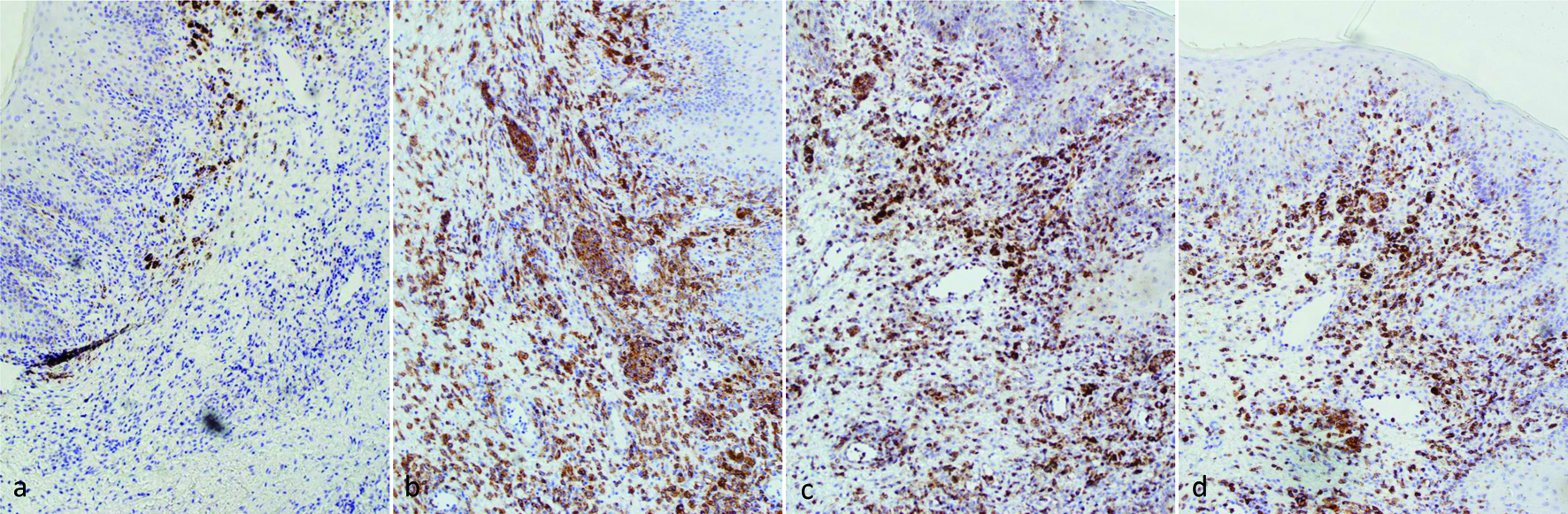 Click for large image | Figure 2. IHC stains of skin biopsy (× 100 magnification): (a) ALK, negative; (b) CD30, positive; (c) CD2, positive; (d): CD5, positive. ALK: anaplastic lymphoma kinase; IHC: immunohistochemistry. |
A subsequent positron emission tomography (PET) scan, conducted a month post-admission, revealed potential reactive lymph nodes linked to the buttock lesion. Despite wound healing post-radiation therapy, new ulcerations appeared and were managed supportively.
Five months after the presentation of initial symptoms, imaging follow-up detected pulmonary nodules and mediastinal lymphadenopathy, among other findings. By the eighth month post-presentation, a CT scan highlighted enlarging pulmonary nodules and a 5.6 cm soft tissue mass situated near the liver.
Ten months after the onset of symptoms, an endoscopic ultrasound (EUS) combined with a fine-needle biopsy (FNA) identified a hypoechoic mass at the head of the pancreas, accompanied by several enlarged lymph nodes surrounding the organ. Based on its location, appearance, and histopathological characteristics, this mass was interpreted as an intra-pancreatic lymphoma invasion, rather than an enlarged lymph node compressing the pancreas. During the course of his treatment and medical consultations, the patient did not display any signs of obstructive jaundice. Additionally, no abnormalities like stenoses or dilatations were detected in either the bile or pancreatic ducts. While pancreatic amylase levels remained consistent within the normal range, a slight uptick in lipase levels was observed, which resolved on its own without intervention.
The pathological examination of the biopsy from the pancreatic head mass revealed the presence of scattered and occasional large, atypical cells amid fibrous tissue (Fig. 3a, b). Notably, discohesive cells intertwined with blood were identified (Fig. 3a, c). Some of these large anaplastic cells exhibited nuclei reminiscent of a “horseshoe” or “wreath”, surrounded by abundant cytoplasm (Fig. 3c). These features are characteristic “hallmark” cells for ALCL. The distinctive morphology of these cells, particularly the eccentrically positioned horseshoe or kidney-shaped nucleus, serves as a unique identifier, setting ALCL apart from other lymphomas.
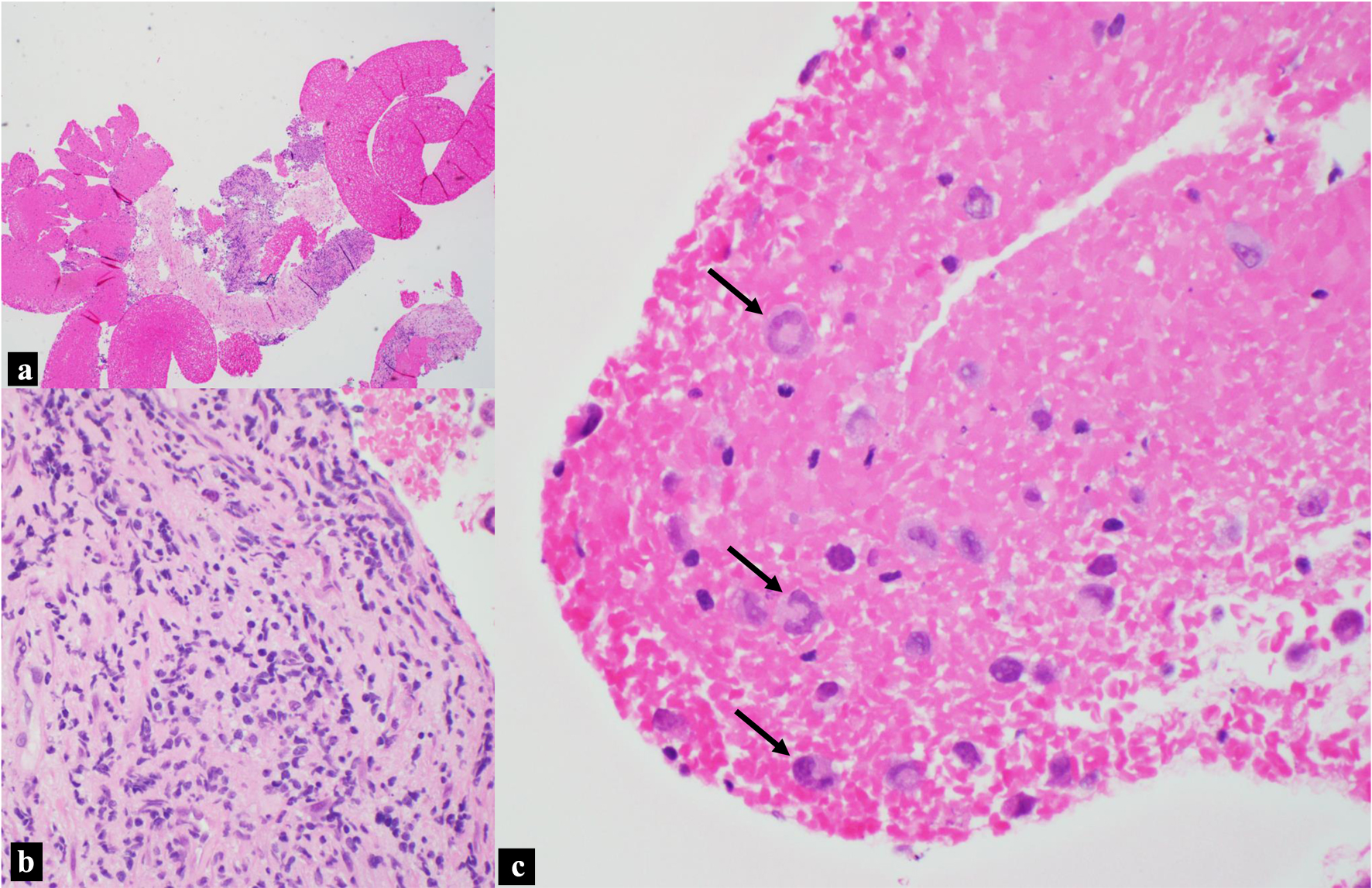 Click for large image | Figure 3. H&E, pancreatic head mass biopsy. (a) Blood and fibrous tissue background with focal hypercellular areas (× 40 magnification). (b) Scattered and rare small clusters of large pleomorphic cells in the background of fibrous tissue (× 400 magnification). (c) Discohesive large anaplastic “hallmark cells” indicated by black arrows, with “wreath” or “horseshoe” nuclei (× 400 magnification). H&E: hematoxylin and eosin. |
IHC staining (Fig. 4) demonstrated that these atypical cells were positive for CD30, CD4, CD43, TIA-1, and MUM-1. They showed subset positivity for CD45 and were negative for markers like CD2, CD3, CD5, CD8, ALK-1, CD20, PAX5, and CD138. Both their morphology and immunophenotype resonated with the characteristics of ALCL. However, it is important to note that peripheral blood flow cytometry was not performed in this case. Nevertheless, the absence of circulating pathologic lymphocytes in the pancreatic FNA underscores the intricacy in designating this condition as a systemic disease.
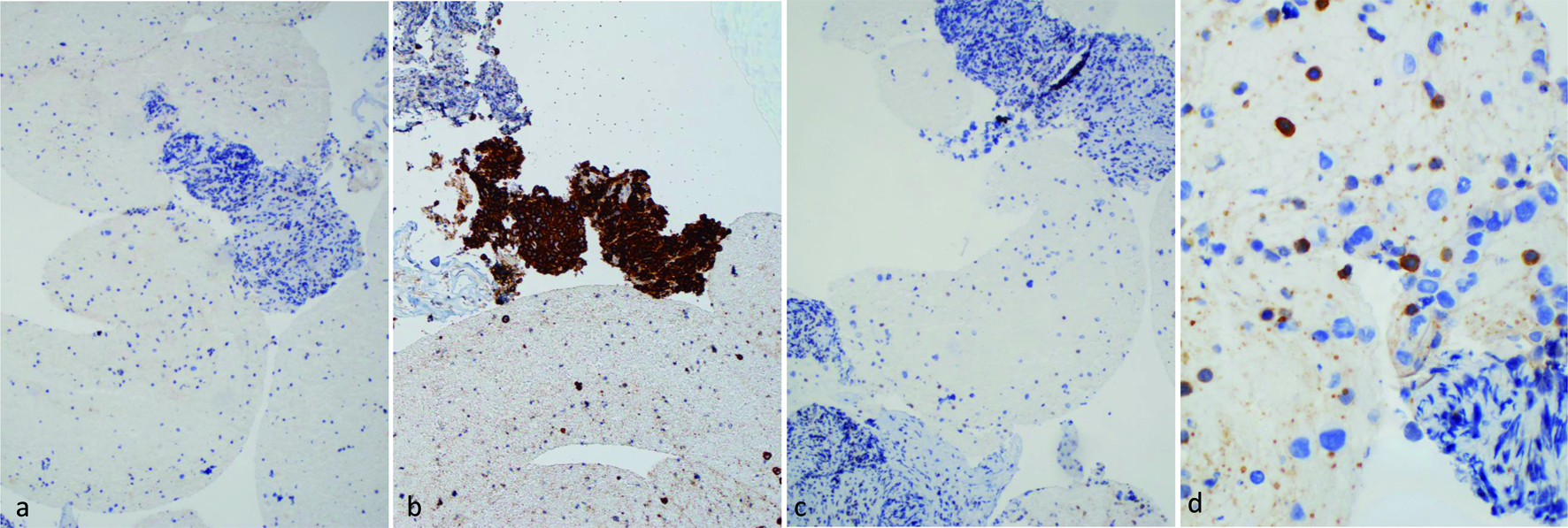 Click for large image | Figure 4. IHC stains of pancreatic mass biopsy (a, b & c × 100 magnification; d × 400 magnification). (a) ALK, negative; (b) CD30, positive; (c) CD3, negative; (d) CD5, negative for large lymphoma cells, with a background of scattered small lymphocytes. ALK: anaplastic lymphoma kinase; IHC: immunohistochemistry. |
Considering the patient’s history of skin manifestations, the differential diagnosis encompassed primary cutaneous ALCL with extracutaneous dissemination and systemic ALK-negative ALCL. However, given the pronounced disease manifestation in the pancreatic head, the existence of pulmonary nodules, widespread lymphadenopathy, and limited skin involvement, a systemic condition was deemed more plausible than a primary cutaneous ailment. Consequently, the patient was conclusively diagnosed with stage IV systemic ALK-negative ALCL.
Eleven months later, a CT scan highlighted a 7 cm mass in the right pelvic region, leading to right ureteral obstruction and hydronephrosis, necessitating hospital readmission. Consequently, the patient was readmitted to the hospital.
The patient was started on three cycles of BV + CHP treatment, which resulted in the resolution of multiple bilateral pulmonary nodules and a significant decrease in the size of the pancreatic head mass from 67.63 to 42.65 mm (Fig. 5). An interval decrease in the size of the soft tissue mass within the right pelvis was also observed. After undergoing another three cycles of BV + CHP, a subsequent PET scan indicated marked progress. The patient continued his HIV treatment with Triumeq, reaching an undetectable HIV viral load within a year, which remained consistent upon subsequent checks. The patient later received an autologous stem cell transplant, achieving a complete response. Two years following chemotherapy, he displayed no signs of lymphadenopathy and has been in remission for over 5 years since the disease’s initial presentation.
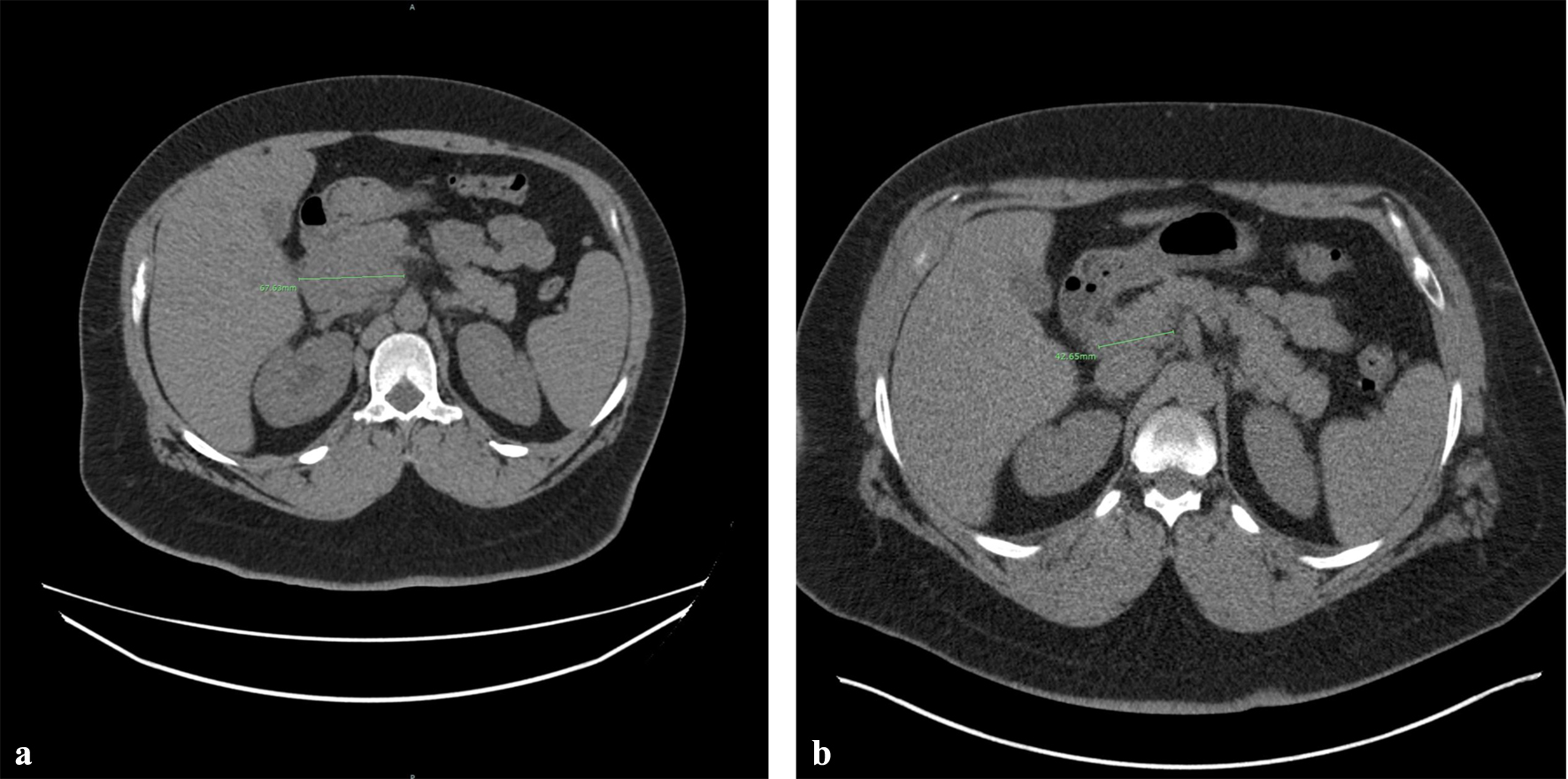 Click for large image | Figure 5. Therapeutic response after three cycles of BV + CHP treatment. CT imaging demonstrates a significant reduction in the size or the pancreatic head mass, with measurements decreasing from 67.63 mm (a) to 42. 65 mm (b). BV + CHP: brentuximab vedotin + cyclophosphamide-doxorubicin-prednisone; CT: computed tomography. |
| Discussion | ▴Top |
The diagnosis of ALK-negative ALCL can be challenging. In this case, the patient presented with an atypical clinical course and an initial lymph node biopsy was inconclusive, and it was only through additional biopsies and comprehensive diagnostic approaches that a definitive diagnosis was made. This highlights the complexity of diagnosing ALCL in the context of atypical presentations and multisite involvement.
Although the initial findings from the buttock ulcer raised the possibility of primary cutaneous ALCL or LyP, the results of subsequent studies including the involvement of multiple extracutaneous sites support the diagnosis of systemic ALK-negative ALCL with cutaneous involvement.
The inconclusive lymph node biopsy results presented an interesting aspect of this case. It is not uncommon to see reactive lymphadenopathy in an HIV-infected patient. Alternatively, the negative result may be due to limited sampling. Dermatopathologist, gastrointestinal (GI) specialists and hematopathologists play a vital role in diagnosing lymphomas, including ALK-negative ALCL. In the context of the GI tract, lymphomas often pose diagnostic challenges due to the diverse clinical and histopathological presentations. The presence of lymphomas in unusual locations, such as the skin and pancreatic head, further complicates the diagnostic process. In these cases, it is essential to pursue alternative diagnostic strategies, such as biopsies from other affected sites or the use of advanced imaging modalities. The success of these alternative strategies may vary depending on the specific lymphoma subtype, emphasizing the need for further research in this area.
IHC staining is crucial in diagnosing ALK-negative ALCL. The unique IHC staining patterns observed between the patient’s buttock ulcer and pancreatic head mass complicated the diagnosis. In the buttock ulcer, the atypical cells showed positivity for CD2 and CD5. In contrast, in the pancreatic head mass, the atypical cells were negative for CD2 and CD5. These variations in IHC staining patterns could be attributed to the evolution of the disease, differences in the tissue microenvironment, or other factors yet to be discovered [8, 9]. Further studies are needed to elucidate the reasons behind these discrepancies and improve diagnostic accuracy.
The rarity of ALK-negative ALCL in HIV-positive patients further complicates the diagnostic process. It is well documented that HIV infection can alter the presentation and behavior of lymphomas [10]. However, the specific impact of HIV on the clinical course of ALK-negative ALCL remains poorly understood. Research focusing on the interplay between HIV infection and ALK-negative ALCL is needed to enhance our understanding of this rare lymphoma subtype and improve diagnostic and therapeutic strategies.
HIV infection is associated with an increased risk of developing lymphoma, particularly non-Hodgkin lymphoma (NHL) [11]. The differential diagnosis of lymphoma in HIV-positive patients is broad and includes a wide range of lymphoproliferative disorders.
The most common subtype of NHL in HIV-positive patients is diffuse large B-cell lymphoma (DLBCL), followed by Burkitt lymphoma and primary central nervous system lymphoma [12]. However, other subtypes of NHL, such as T-cell lymphomas, and primary effusion lymphoma, have also been reported in this population [11]. In addition to NHL, HIV-positive patients are also at increased risk of developing Hodgkin lymphoma (HL), with an incidence rate that is three to five times higher than in the general population [12]. The most common subtype of HL in HIV-positive patients is mixed cellularity, followed by nodular sclerosis and lymphocyte-rich [13]. The occurrence of HL is less frequent in cases of severe immunosuppression compared to moderate immunosuppression. The rise in HL incidence among individuals with HIV/acquired immune deficiency syndrome (AIDS) since 1996 can likely be attributed to the enhancement in CD4 counts brought about by the use of highly active antiretroviral therapy (HAART) [13].
Recent studies have suggested that ALK-negative ALCL may be more common than previously thought and is associated with a poorer prognosis compared to ALK-positive ALCL [14]. In addition, it has been suggested that ALK-negative ALCL is a more heterogeneous disease entity, with a higher frequency of genetic alterations and mutations compared to ALK-positive ALCL [15]. For example, rearrangements involving the DUSP22 and IRF4 genes have been found in approximately 30-40% of cases of systemic ALK-negative ALCL and may be associated with a more favorable prognosis [14-16]. Conversely, various factors have been associated with a less favorable prognosis, including rearrangements in the TP63 gene [17], a deficiency of P53 [18], the individual or combined expression of p53 and β-catenin [19], as well as an increased expression of interleukin-2 receptor α (IL-2Rα) [20].
These molecular alterations may also have implications for the development of targeted therapies for ALCL. Unfortunately, these cytogenetic and molecular tests were not performed in our case due to limited tissue.
In summary, this case report underscores the diagnostic challenges associated with ALK-negative ALCL, particularly in an HIV-positive patient. The inconclusive lymph node biopsy results and the involvement of unusual sites, such as the pancreatic head, emphasize the need for thorough investigation and a high index of suspicion. The difficulties encountered during the diagnostic process for ALK-negative ALCL may be attributed to its rarity, as well as the diverse clinical presentations it can manifest. Further research is needed to better understand the molecular underpinnings of ALK-negative ALCL, the impact of HIV infection on its presentation, and to improve diagnostic accuracy and therapeutic strategies for this rare lymphoma subtype.
Conclusion
This captivating case report illuminates the intricate diagnostic challenges that arise when dealing with the exceedingly rare ALK-negative ALCL, especially in the context of HIV infection. Importantly, clinicians must remain alert to the possibility of multisite involvement, as seen in this case with involvement of the pancreas, emphasizing the need for a comprehensive diagnostic workup. A robust panel of immunohistochemical markers, including CD30, CD4, CD8, CD3, ALK-1, and others, should be employed to aid in accurate diagnosis. The impressive response to the targeted BV + CHP therapy administered in this case exemplifies the potential success of tailored treatment approaches for ALCL patients. Yet, further investigation into the molecular mechanisms underpinning ALCL and advancements in diagnostic precision and therapeutic modalities are needed to enhance patient outcomes in this rare lymphoma subtype.
Acknowledgments
We would like to thank the dermatopathologist, GI pathologist, clinicians, radiologists, and histotechnicians for their contributions in diagnosing and managing the patient’s ALK-negative anaplastic large cell lymphoma. We also thank the patient for his participation in this case report.
Financial Disclosure
This work has not received any form of financial support or funding.
Conflict of Interest
The authors declare no conflict of interest.
Informed Consent
Informed consent was obtained from the patient for the publication of this case report and any accompanying images.
Author Contributions
All authors contributed significantly to the conception, design, and interpretation of data. HHH drafted the manuscript. XQ provided critical revisions and approved the final version of the manuscript. Both authors have read and approved the final manuscript.
Data Availability
The data used to support the findings of this study are available from the corresponding author upon request.
| References | ▴Top |
- Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, Advani R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375-2390.
doi pubmed pmc - Alaggio R, Amador C, Anagnostopoulos I, Attygalle AD, Araujo IBO, Berti E, Bhagat G, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia. 2022;36(7):1720-1748.
doi pubmed pmc - Parrilla Castellar ER, Jaffe ES, Said JW, Swerdlow SH, Ketterling RP, Knudson RA, Sidhu JS, et al. ALK-negative anaplastic large cell lymphoma is a genetically heterogeneous disease with widely disparate clinical outcomes. Blood. 2014;124(9):1473-1480.
doi pubmed pmc - Nagtegaal ID, Odze RD, Klimstra D, Paradis V, Rugge M, Schirmacher P, Washington KM, et al. The 2019 WHO classification of tumours of the digestive system. Histopathology. 2020;76(2):182-188.
doi pubmed pmc - Savopoulos CG, Tsesmeli NE, Kaiafa GD, Zantidis AT, Bobos MT, Hatzitolios AI, Papavramidis ST, et al. Primary pancreatic anaplastic large cell lymphoma, ALK negative: a case report. World J Gastroenterol. 2005;11(39):6221-6224.
doi pubmed pmc - Hughes B, Habib N, Chuang KY. Primary anaplastic large cell lymphoma of the pancreas. ACG Case Rep J. 2019;6(10):e00231.
doi pubmed pmc - Stein H, Foss HD, Durkop H, Marafioti T, Delsol G, Pulford K, Pileri S, et al. CD30(+) anaplastic large cell lymphoma: a review of its histopathologic, genetic, and clinical features. Blood. 2000;96(12):3681-3695.
pubmed - Benharroch D, Meguerian-Bedoyan Z, Lamant L, Amin C, Brugieres L, Terrier-Lacombe MJ, Haralambieva E, et al. ALK-positive lymphoma: a single disease with a broad spectrum of morphology. Blood. 1998;91(6):2076-2084.
pubmed - Grogg KL, Miller RF, Dogan A. HIV infection and lymphoma. J Clin Pathol. 2007;60(12):1365-1372.
doi pubmed pmc - Powles T, Thirlwell C, Nelson M, Bower M. Immune reconstitution inflammatory syndrome mimicking relapse of AIDS related lymphoma in patients with HIV 1 infection. Leuk Lymphoma. 2003;44(8):1417-1419.
doi pubmed - Biggar RJ, Engels EA, Frisch M, Goedert JJ, AIDS Cancer Match Registry Study Group. Risk of T-cell lymphomas in persons with AIDS. J Acquir Immune Defic Syndr. 2001;26(4):371-376.
pubmed - Navarro JT, Molto J, Tapia G, Ribera JM. Hodgkin lymphoma in people living with HIV. Cancers (Basel). 2021;13(17):4366.
doi pubmed pmc - Biggar RJ, Jaffe ES, Goedert JJ, Chaturvedi A, Pfeiffer R, Engels EA. Hodgkin lymphoma and immunodeficiency in persons with HIV/AIDS. Blood. 2006;108(12):3786-3791.
doi pubmed pmc - Savage KJ, Harris NL, Vose JM, Ullrich F, Jaffe ES, Connors JM, Rimsza L, et al. ALK- anaplastic large-cell lymphoma is clinically and immunophenotypically different from both ALK+ ALCL and peripheral T-cell lymphoma, not otherwise specified: report from the International Peripheral T-Cell Lymphoma Project. Blood. 2008;111(12):5496-5504.
doi pubmed - Parkhi M, Bal A, Das A, Kashyap D, Bhardwaj S, Prakash G, Malhotra P. ALK-negative anaplastic large cell lymphoma (ALCL): prognostic implications of molecular subtyping and JAK-STAT pathway. Appl Immunohistochem Mol Morphol. 2021;29(9):648-656.
doi pubmed - Pina-Oviedo S, Ortiz-Hidalgo C, Carballo-Zarate AA, Zarate-Osorno A. ALK-negative anaplastic large cell lymphoma: current concepts and molecular pathogenesis of a heterogeneous group of large T-Cell lymphomas. Cancers (Basel). 2021;13(18):4667.
doi pubmed pmc - Boi M, Rinaldi A, Kwee I, Bonetti P, Todaro M, Tabbo F, Piva R, et al. PRDM1/BLIMP1 is commonly inactivated in anaplastic large T-cell lymphoma. Blood. 2013;122(15):2683-2693.
doi pubmed - Lobello C, Tichy B, Bystry V, Radova L, Filip D, Mraz M, Montes-Mojarro IA, et al. STAT3 and TP53 mutations associate with poor prognosis in anaplastic large cell lymphoma. Leukemia. 2021;35(5):1500-1505.
doi pubmed pmc - Richardson AI, Yin CC, Cui W, Li N, Medeiros LJ, Li L, Zhang D. p53 and beta-catenin expression predict poorer prognosis in patients with anaplastic large-cell lymphoma. Clin Lymphoma Myeloma Leuk. 2019;19(7):e385-e392.
doi pubmed - Liang HC, Costanza M, Prutsch N, Zimmerman MW, Gurnhofer E, Montes-Mojarro IA, Abraham BJ, et al. Super-enhancer-based identification of a BATF3/IL-2R-module reveals vulnerabilities in anaplastic large cell lymphoma. Nat Commun. 2021;12(1):5577.
doi pubmed pmc
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.