

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://www.thejh.org |
Case Report
Volume 13, Number 3, June 2024, pages 108-115

Characterization and Clinical Assessment of a Peculiar Case of Hemolytic Anemia
Fulvio Castelgrandea, e, Gemma Violab, e, Cinzia Calabresea, e, Mariannina Iozzob, Renato Massoudb, c, Massimo Pierib, c, Marilena Minierib, c, Gaspare Adornod, Sergio Bernardinib, c, Alessandro Terrinonib, c, f
aSchool of Laboratory Medicine and Pathology, University of Rome Tor Vergata, Via Cracovia, 00133 Rome, Italy
bLaboratory Medicine Department, University Hospital of Tor Vergata, Viale Oxford, 1-00133 Rome, Italy
cDepartment of Experimental Medicine, University of Rome Tor Vergata, Via Montpellier, 1-00133 Rome, Italy
dDepartment of Biomedicine and Prevention, University of Rome Tor Vergata, Via Montpellier, 1-00133 Rome, Italy
eThese three authors contributed to the study equally.
fCorresponding Author: Alessandro Terrinoni, Laboratory Medicine Department, University Hospital of Tor Vergata, Viale Oxford, 1-00133 Rome, Italy
Manuscript submitted October 12, 2023, accepted January 5, 2024, published online June 28, 2024
Short title: A Peculiar Case of Hemolytic Anemia
doi: https://doi.org/10.14740/jh1204
| Abstract | ▴Top |
Thalassemic diseases are characterized by a reduced (β+) or absent (β0) synthesis of the globin chains of hemoglobin (Hb) due to genetic mutations. β-thalassemia was more frequent in the Mediterranean area, but now it is diffused worldwide. Three possible genetic forms can be distinguished: β0/β0, the most severe (Cooley’s disease); β0/β+ of intermediate severity; β+/β+ associated with β-thalassemia intermedia or minor. Recently, a clinical non-genetic classification has been proposed: transfusion-dependent thalassemia (TDT), requiring regular lifetime blood transfusions, and non-transfusion-dependent thalassemia (NTDT), requiring occasional transfusions to manage acute cases. In this report, we studied a patient whose blood count indicated a severe anemia but also showed thrombocytosis, leukocytosis, and an elevated number of nucleated red blood cells (NRBC). These altered blood parameters suggested initially a possible diagnosis of hemoglobinopathy or myeloproliferative syndrome. The molecular and genetic analyses demonstrated the presence of HbF (5.3%) and HbA2 (7.7%) and the presence of the homozygote mutation (IVS1.6T>C) in the β-globin gene. According to these data, a diagnosis of β-thalassemia intermedia form has been proposed. Nevertheless, the clinical condition, the presence of thrombocytosis, leukocytosis, an elevated number of NRBC, and the frequent blood transfusions lead to reclassification of the patient as TDT subject. Consequently, this result suggests that a unique genotype-phenotype correlation is not possible in the presence of β+mutations since other concomitant pathologies can exacerbate the disease.
Keywords: β-thalassemia; Transfusion-dependent thalassemia; Anemia; Hemoglobin
| Introduction | ▴Top |
Thalassemias are a group of inherited hematological disorders caused by defects in the synthesis and/or expression of one or more hemoglobin (Hb) chains. α-thalassemia is caused by reduced or absent synthesis of α-globin chains [1]; β-thalassemia is caused by reduced or absent synthesis of β-globin chains [2]. Low expression or absence of the specific chain can be determined by large deletions or mutations that introduce a stop codon.
α-thalassemia is highly prevalent in all tropical and subtropical regions, especially in the equatorial region of Africa [3]. Intermediate and severe forms of α-thalassemia are very rare in North America and Europe and are mostly observed in populations migrating from Southeast Asia or Mediterranean countries [4]. α-thalassemia is often caused by deletions of one or both alleles (HBA1 and HBA2), and the severity of the clinical picture correlates with α-globin deficiency. Several clinical subtypes of increasing severity are distinguished: silent α-thalassemia (-/α α/α), α-thalassemic trait (-/- α/α or -/α -/α), hemoglobinopathy H (HbH) (-/- -/α) and Bart’s Hb hydrops fetalis (-/- -/-) [2].
β-thalassemia is typical of the Mediterranean basin, but epidemiological data show that severe forms are present also in Central and South-East Asia, India and China [4]. Migration phenomena are generally recognized as the cause of the global distribution of the disease [5], although there is still a “founder” effect that localizes the disease more in certain regions or in populations that have a high degree of inbreeding for ethnic-religious reasons [6]. β-thalassemia is caused by point mutations introducing stop codons, splicing variants or deletions in the β-globin gene. The result is a reduction in β-globin protein in β+ mutations, whereas β0 mutations completely abolish protein synthesis. Therefore, there are three possible genotypes/phenotypes, of which β0/β0 (Cooley’s disease) is the most severe. It is characterized by the absence of β-chains, resulting in the absence of HbA1 and an increase in HbF. The β0/β+, with an intermediate severity, is characterized by partial deficiency of the β-chains, and the β+/β+ variant, which may be associated with β-thalassemia intermedia or β-thalassemia minor [6].
Recently, patients have also been clinically classified according to their transfusion needs. The subjects requiring regular blood transfusions have been classified in the transfusion-dependent thalassemia (TDT) group. The patients requiring occasional transfusions to manage acute events that cause rapid reduction in Hb levels have been included in the non-transfusion-dependent thalassemia (NTDT) group [5]. NTDT phenotypes include β-thalassemia intermedia, mild/moderate β-thalassemia with HbE, and hemoglobinopathy H [7]. TDT phenotypes include β-thalassemia major and severe forms of β-thalassemia with HbE [6, 8].
In TDT patients, ferro-chelating therapy is necessary to prevent morbidity and mortality due to iron overload that develops because of the frequent transfusions required to manage the severe anemia resulting from ineffective erythropoiesis [5].
Ineffective erythropoiesis (IE) is a particular form of anemia in which an increase in erythroid cells is not matched by an increase of red blood cell (RBC) [9]. Erythropoiesis needs iron to ensure an efficient Hb synthesis and RBC differentiation, thus, under anemia or hypoxia conditions an increase in iron uptake is generally promoted [10]. One of the models explaining the increase of iron in hypoxic or anemic conditions is the suppression of hepcidin [11]. Hepcidin is a protein synthesized in the liver; it blocks intestinal iron absorption and release by macrophages. Hepcidin performs its function by binding to ferroportin, a protein that regulates iron release from intestinal mucosal cells and macrophages. The binding of hepcidin to ferroportin prevents the latter from functioning thus blocking the release of iron into the blood [11, 12]. In IE, erythroferrone, one of the main hormones regulating erythropoiesis, reduces hepcidin synthesis causing an increase of circulating iron, which, instead of being used for Hb synthesis, is deposited in organs, damaging them [13].
In β-thalassemia, the normal balance of α-globin and β-globin chains is lost, thus the excess of free α-globin chains bind heme forming insoluble toxic aggregates called hemichromes, which precipitate and damage the RBC [14].
In individuals with β-thalassemia, damaged RBCs are more easily sequestered by the spleen, which in turn provides their elimination (eryptosis). As the disease progresses, IE and the resulting anemia exacerbate extramedullary erythropoiesis in spleen and liver. The resulting splenomegaly is further worsened by eryptosis with the increase of anemia and hypoxia [10]. This condition is dangerous both for the risk of organ failure and because patients with enlarged spleen require more frequent blood transfusions to survive. Therefore, splenectomy becomes necessary due to the excessive enlargement of the organ. However, this condition would reduce the patient’s quality of life for an increased susceptibility to infections and thrombotic phenomena [10, 14]. In this report, we analyzed a thalassemic patient who, according to the genetic classification, should be included in the β-thalassemia intermedia group. However, the clinical conditions and the request of continuous transfusions result in the inclusion of the patients in the more severe TDT group. In this study, we have demonstrated that a unique genotype-phenotype correlation is not possible in presence of the known β+ mutations. The laboratory diagnosis was carried out on two main levels: the first level involved complete blood count, morphological evaluation of erythrocyte, biochemical investigations (bilirubin, iron, ferritin assays), electrophoresis and high-performance liquid chromatography (HPLC) chromatography of the Hb molecules, while the second level of investigation was aimed at dissecting the molecular defect of DNA [8, 15].
| Case Report | ▴Top |
Investigations
We analyzed a 39-year-old female patient admitted to the Emergency Department of University Hospital of Tor Vergata (Rome, Italy) for the presence of bilateral declivous edema. The patient arrived from Lebanon via a humanitarian corridor with a severe language barrier. Her medical history documented a previous severe anemia not well specified, thrombocytosis, leukocytosis, and hepatomegaly. A splenectomy was also discovered during clinical evaluation of the patient, and she underwent the procedure in 2012. The patient also reported needing regular transfusion therapy for severe anemia, with the last transfusion performed 10 days before access to our Emergency Department with the onset of transfusion reaction.
The clinical and laboratory data were consistent with either a myeloproliferative syndrome or a hemoglobinopathy.
Diagnosis
Once hematological samples were received in the laboratory, a complete blood count was performed, which showed severe anemia with Hb value of 4.7 g/dL; mean corpuscular volume (MCV) and mean corpuscular hemoglobin (MCH) equal to 79 fL and 23.8 pg, respectively; red blood cell distribution width-coefficient of variation (RDW-CV) and red blood cell distribution width-standard deviation (RDW-SD) significantly increased, index of a high degree of erythrocyte anisopoikilocytosis; platelets equal to 980 × 103/µL, calculated with impedance method (PLT-I) and then confirmed by the optical method (PLT-O: 1,208 × 103/µL); leukocytosis (24.51 × 103/µL of white blood cell (WBC)); neutrophils 36% and lymphocytes 55.4% (Fig. 1a, b); significant presence of nucleated red blood cells (NRBC) (45.6%) (Fig. 2a, b; purple). Blood count has been performed using the MINDRAY BC 6800 Plus system. It provides complete blood count, differential leukocyte counts in five parts, measurement of Hb concentration and measurement of reticulocytes and NRBC in blood samples, as well as analysis of body fluids; for NRBC, the hematological analyzer performs a direct count and derives the percentage.
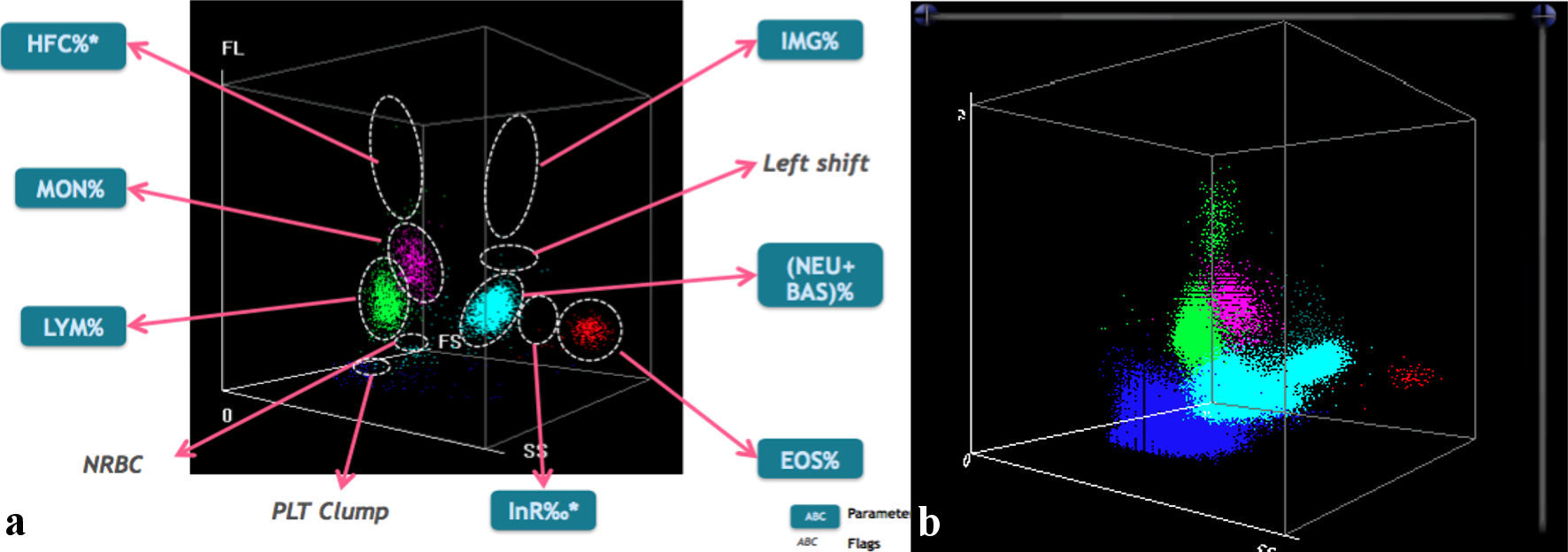 Click for large image | Figure 1. (a) In the left image, the typical scattergram showing the distribution of different white blood cell populations in three-dimensional space (by MINDRAY BC 6800). (b) In the right image, the patient graph shows clear lymphocytosis (green population). The presence of high-fluorescent cells (HFCs) is indicative of the probable presence of blasts or atypical lymphocytes. In addition, there is a nonhomogeneous cloud of nucleated red blood cells (NRBCs), light blue color) located between the population of lymphocytes (LYM) and neutrophils (NEU). At the bottom, blue in color, is the expanded cloud of platelets (PLT), an obvious sign of thrombocytosis. EOS: eosinophils; MON: monocytes; BAS: basophils. |
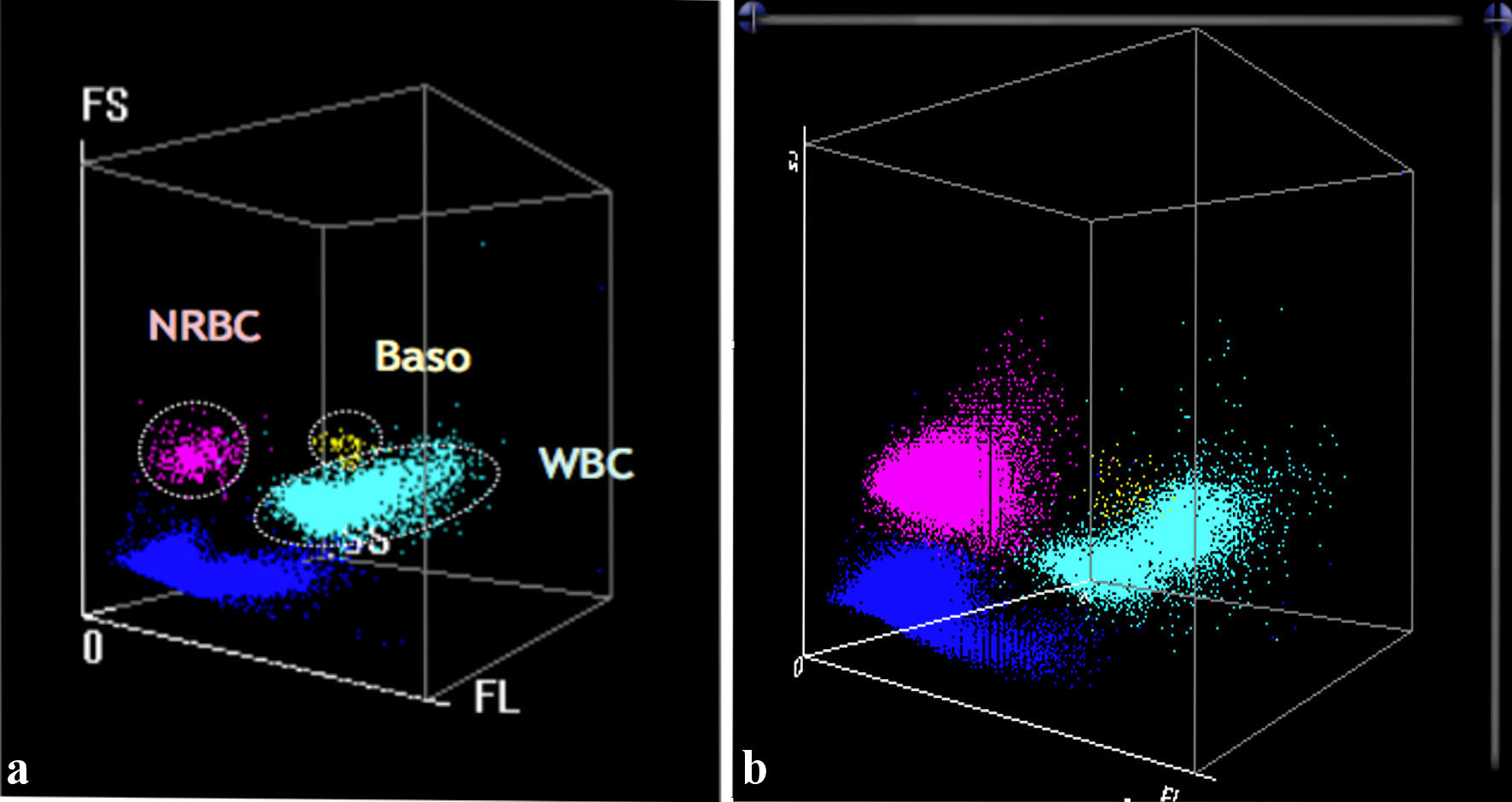 Click for large image | Figure 2. (a) In the left image, the WNB channel (MINDRAY BC 6800) is represented, which is dedicated exclusively to NRBCs (purple), basophils (Baso) (yellow) and neutrophils (light blue). (b) The patient graph (right) confirms the presence of a discrete population of NRBCs compared with the reference graphical pattern (left). WNB: white cell nucleated basophil channel; NRBCs: nucleated red blood cells; WBC: white blood cell. |
The hepatic function tests (albumin, aspartate aminotransferase (AST), alanine aminotransferase (ALT), γ-glutamyl transferase (GGT)) and renal function (electrolytes, creatinine, and azotemia) were in the normal range, but an increase in total bilirubin (2 mg/dL, reference range: 0.3 - 1 mg/dL) and direct bilirubin (0.79 mg/dL, reference range: 0.1 - 0.3 mg/dL) levels was reported, indicating an impairment of the liver function. The ferritin dosage showed an increase equal to 2,980 ng/mL, while transferrin was decreased (166 mg/dL). These biochemical parameters have been performed by Alinity-c system (Abbott).
The direct Coombs’ test (performed using standard methods), for the detection of anti-erythrocyte antibodies, yielded positive detection of anti-total, anti-immunoglobulin (Ig)G and anti-C antibodies. This is likely a result of a transfusion (probably the last one) of an incompatible unit of transfused blood, thus confirming what the patient previously reported.
Microbiological tests were also carried out, which excluded viral infections (hepatitis B virus (HBV), hepatitis C virus (HCV), human immunodeficiency virus (HIV), varicella-zoster virus (VZV) and cytomegalovirus (CMV)), while positive results were found for previous herpes simplex virus (HSV) type 1 and Epstein-Barr virus (EBV) infection (EBV nuclear antigen (EBNA) IgG and viral capsid antigen (VCA) IgG). However, the previous splenectomy, in the first instance, could justify the thrombocytosis and leukocytosis of the patient, but onco-hematologic investigations has been performed to exclude the suspicion of positivity for a JAK2 myeloproliferative neoplasm. For this analysis, we obtained negative results both for clone EPN (paroxysmal nocturnal hemoglobinuria) and for the search of the V617F mutation of the JAK2 gene by DNA extraction and amplification by real-time polymerase chain reaction (PCR), and allele-discrimination analysis.
The analysis of the coagulation has been also carried out due to the referred personal and family history of venous thrombosis. The results showed the presence of the MTHFR A1298C mutation in homozygosis, but with homocysteine levels in the normal range (3.9 µmol/L), absence of mutation of factor V Leiden, absence of mutation of factor II (prothrombin), protein S and C were both within the normal range. The analysis of protein C was carried out with the chromogenic method and protein S with the immunoturbidimetric method, using the StaGo CoagOne.
Considering the complexity of the hematological findings (double hemolytic component, hemoglobinopathy and autoimmune, the etiology of which had to be determined) and to investigate the pathogenetic nature of hemolytic anemia, a peripheral venous smear and Hb electrophoresis were performed.
The peripheral blood smear revealed marked RBC poikilocytosis with numerous target RBCs, dacrocytes and schistocytes (Fig. 3), the genesis of which was most likely attributable to the autoimmune hemolytic process triggered by the presence of anti-erythrocyte antibodies (anti-T, anti-IgG and anti-C), highlighted on the previous direct Coombs’ test.
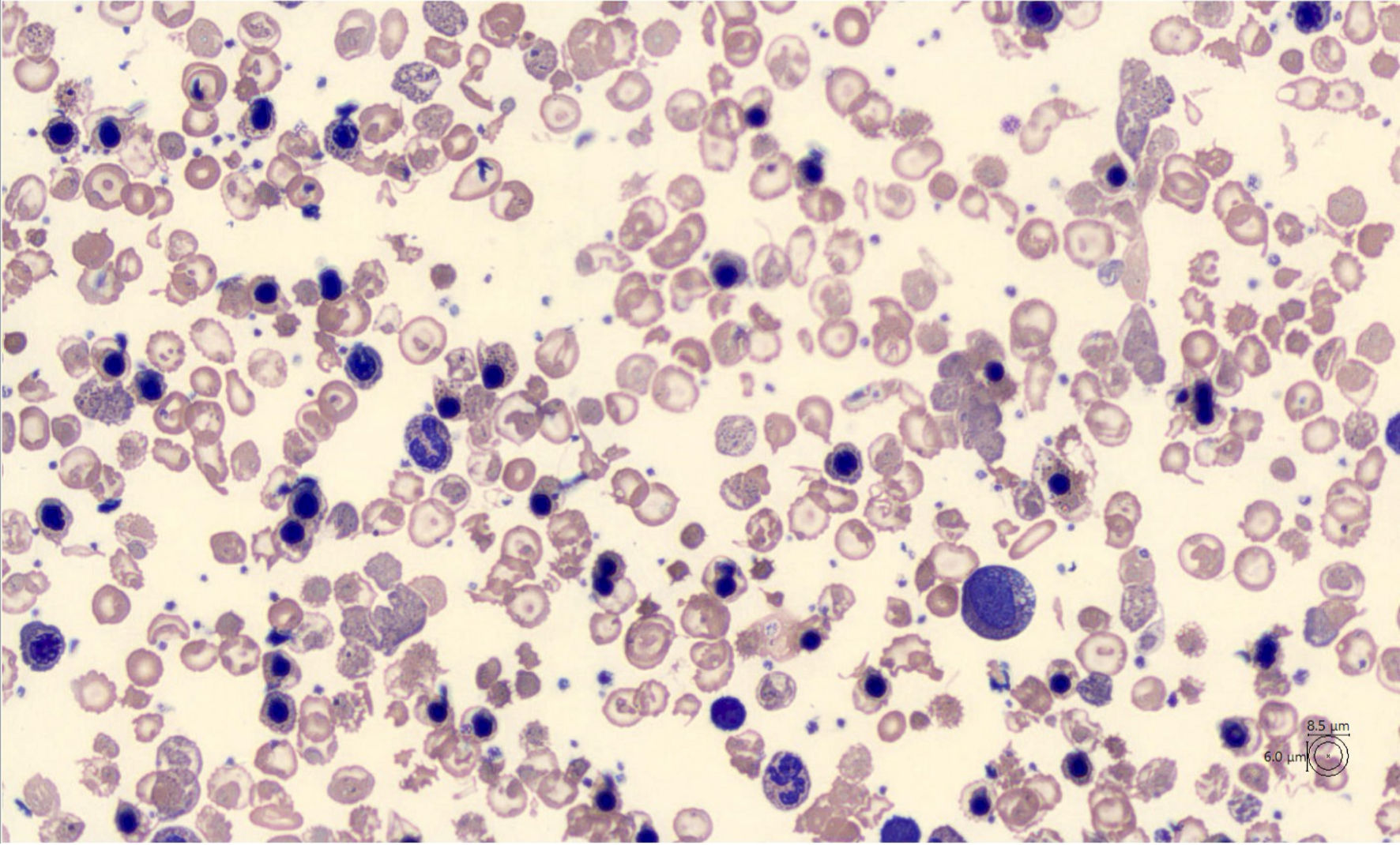 Click for large image | Figure 3. Peripheral blood smear (CellaVisions) showing marked anisopoikilocytosis of red blood cells (RBC) and the presence of erythroblasts predominantly in orthochromatic phase. The population of RBC consists mainly of schistocytes, dacrocytes, target cells and acanthocytes. |
Furthermore, marked anisocytosis characterized by numerous red cells with basophilic punctuation was also evident, the presence of Howell-Jolly bodies, erythroblastosis with dimorphism, marked anisochromia and polychromasia, reticulocytosis and several giant thrombocytes without agglutinates were also present. Moreover, the peripheral blood smear showed the white series accounts for 40% of neutrophils, some of which hyper-granulated, the presence of myelocytes and metamyelocytes, 2.8% and 0.9%, respectively, lymphocytes equal to 50.8% some of which activated, monocytes equal to 5.5% and basophils equal to 0% (Fig. 3). These results together suggested a possible diagnosis of a type of thalassemia. The morphological analysis has been performed using the MINDRAY SC-120 automatic slide preparer to perform the peripheral blood smear. The smear, stained using the May Grunwald-Giemsa panoptic method, was acquired, and processed by the image analyzer CellaVision DM1200, which provides a WBC count of 200 cells.
Given the general hematological results, a chromatographic analysis was also performed showing an increase in HbF (5.3%) and HbA2 (7.7%) (Fig. 4). Chromatographic analysis of Hb by HPLC method was performed by the TOSOH Hlc-723 G11 instrument.
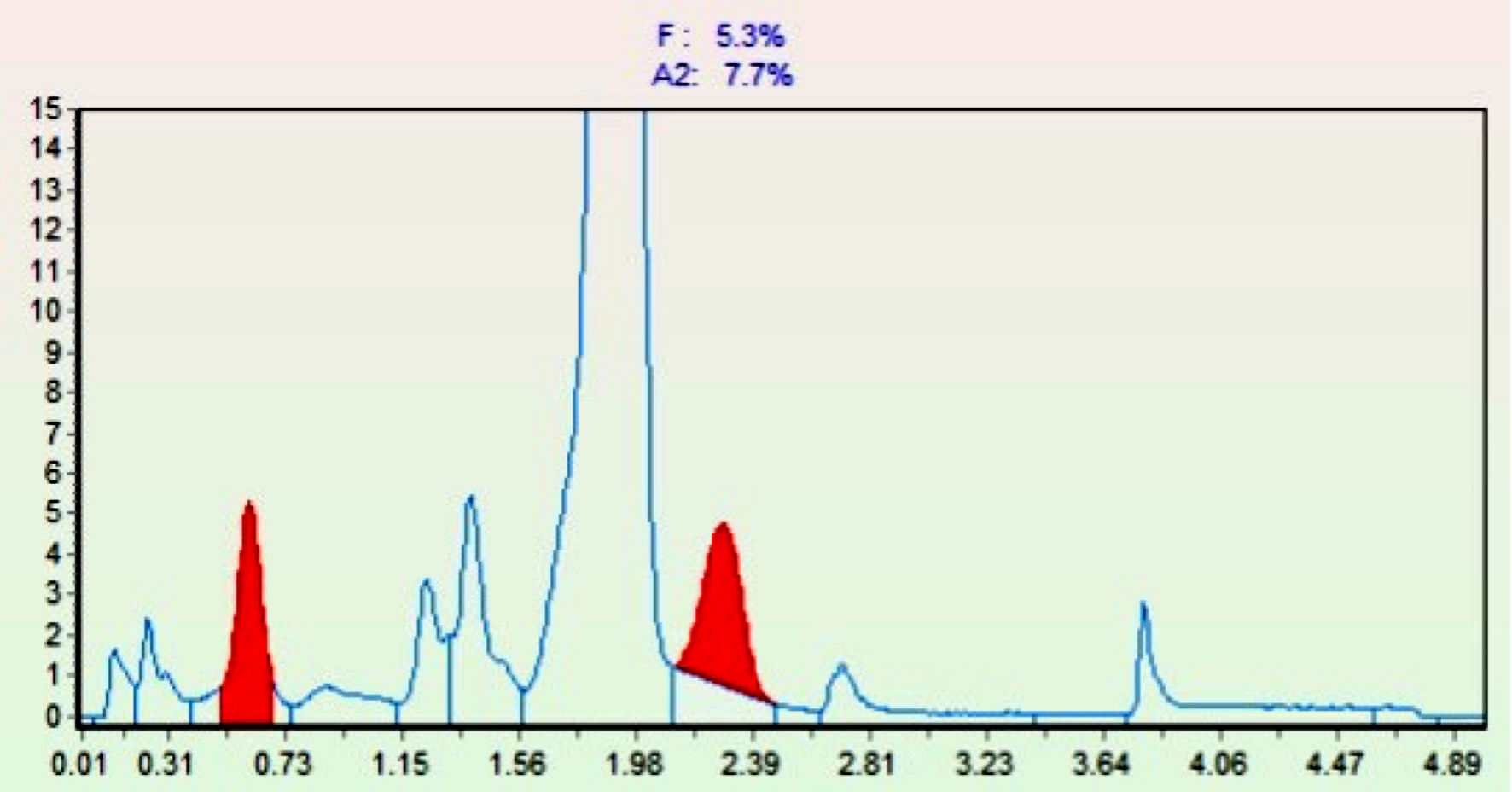 Click for large image | Figure 4. Chromatogram of pathological hemoglobins. On the abscissa (X-axis), retention times are reported, while on the ordinate (Y-axis), the percentages of the different hemoglobins are indicated. The red peaks show the increase in HbF (first red peak) and HbA2 (second red peak). Hb: hemoglobin. |
The chromatographic data of Hb distribution were suggestive of a possible form of β-thalassemia. To confirm this hypothesis, a genetic re-evaluation with sequencing for the β-globin gene was performed for the presence of possible mutations or deletions.
The gene sequencing of β-globin (HBB) gene, by Sanger sequencing using ABI-3500, showed the presence in homozygosis of the β+ pathogenic variant HGVS:NG_059281.1(NM_000518.5): c.92+6T>C (IVS1.6T>C) of the gene, determining the genotype as HGVS:NG_059281.1(NM_000518.5): c.92+6T>C(;)(92+6T>C). This is a nonsense mutation that introduces a stop to codon 37 of the gene. This mutation, already described in the literature, is associated, when carried as homozygous, with different degrees of thalassemia [16], generally intermedia. This result, associated with morphological hematological results and chromatographic analysis, could suggest a diagnosis of β-thalassemia intermedia.
To exclude organ damage due to hemosiderin deposition, due to the numerous transfusions and to the high serum ferritin values, and to further exclude the presence of solid tumors as hematological indication, a total-body computed tomography (CT) scan with contrast was also performed. The CT results showed an enlarged liver, the presence of multiple and voluminous hypodense formations in the probable site of the left adnexa, the largest of which had a parietal calcified component measuring 56 × 42 mm. In the inguinal region, lymph node formations of increased size were reported, with maximum axial dimensions of approximately 20 × 9 mm on the left and 20 × 9 mm on the right. In the skeletal context, at the level of the vertebral somata and the pelvic bones, multiple areas of bone rarefaction were observed. Considering the CT scan results and the corticosteroid therapy, which the patient was taking, it was considered appropriate to perform a femoral and lumbar dual-energy X-ray absorptiometry (DEXA), which resulted in a global T-score of -1.8, consistent with osteopenia and a moderate risk of fracture.
In addition, to complete the diagnostic procedure, an osteo-medullary bone biopsy was performed, which showed a hypercellular bone marrow with the presence of 90% of erythroblasts, mainly in the orthochromatic phase with numerous Howell-Jolly bodies, and in the absence of blasts.
No growth retardation, mainly associated to α-thalassemia [17] and thalassemic faces has been reported in our patient.
Treatment
During the hospitalization period, the patient received steroids, gastroprotective, ferro-chelating, hypokalemic, prophylactic anticoagulant therapy to contrast the increased thrombotic risk due to the thrombocytosis followed to the splenectomy.
For the severe anemia treatment, the patient underwent transfusion of two bags of prefiltered packed red blood cells to be infused in slow teardrop. The main blood count values at admission, just after transfusion, and at the dismission time, are shown in Table 1.
 Click to view | Table 1. Blood Count Pre- and Post-Transfusion |
| Discussion | ▴Top |
β-thalassemia is a hereditary and heterogeneous group of diseases caused by several point or complex deletion/insertion mutation in the β-globin gene, which tendentially introduce stop codons, splicing variants or important modifications, resulting in a reduced or no production of β-globin chains [2]. Excess α-chains provokes damage to RBCs, which are more easily sequestered by the spleen and destroyed. This phenomenon of IE can lead to a clinical picture of severe anemia, and erythroid hyperplasia with bone marrow expansion (then hepatosplenomegaly) [18]. In the case described in this paper, the scattergram of the blood count of the patient, as well as indicating severe anemia, also showed thrombocytosis and leukocytosis with presence of a high number of NRBC (Figs. 1, 2). Therefore, the clinical picture could be suggestive of both hemoglobinopathy and myeloproliferative syndrome. Further morphological examination described in result section oriented towards the diagnosis of hemoglobinopathy, then confirmed with globin chromatographic analysis and DNA sequencing. Biochemical analyses and onco-hematological investigations have excluded the possibility of a positive JAK2 myeloproliferative syndrome, even in the presence of leukocytosis. As reported in literature, the myelodysplastic syndrome can be associated with α-thalassemia, due to mutation in ATRX gene, but it is also associated with mental retardation [19], which is not reported in our case. Rare reports describing the coexistence in patients of myelodysplastic syndrome with β-thalassemia intermedia have been described [20], but with no genetic association. Considering the data obtained, a final diagnosis of β-thalassemia intermedia has been established.
The mutation found that the NM_000518.5: c.92+6T>C (IVS1.6T>C) is widespread in West Bank populations, with a relatively high frequency of 11.3% [16]. From the analysis of frequency and distribution, it is believed that it could be dated back to very ancient times and supports the hypothesis that it originated in this region [21]. The geographical origin of our patient (Lebanon) is very close to the mutation origin area, confirming the diffusion of the mutation from its origin point. Moreover, compatibly with literature data indicating a variable clinical phenotype, even if our patient is placed in the clinical window of β-thalassemia intermedia, it is interesting to note that the clinical findings point to a more severe type of thalassemia.
The first-line therapy of patients with β-thalassemia intermedia consists of occasional blood transfusions to manage moderate/severe anemia in combination with ferro-chelating therapy to limit iron accumulation. Moreover, as in the case described in this paper, anticoagulant drugs as adjuvant therapy are used to prevent thrombotic events in splenectomized patients. Initially, the gold standard therapy was considered the administration of vitamin K antagonists, which in turn required frequent blood samples to monitor international normalized ratio (INR) and thus more venous accesses. Recently, the use of direct oral anticoagulants (DOACs), which exert their anticoagulant action by inhibiting a specific factor of the coagulation cascade is preferred, inducing, compared with anti-vitamin K drugs, a more stable anticoagulation action without the need for continuous INR monitoring and consequent reduction of venous accesses [12, 22].
New recent therapies are emerging in the treatment of β-thalassemia-like patients, which consists of allogeneic bone marrow transplantation in severe anemia patients (a sort of gene therapy) and aimed to eliminate continuous transfusions. However, there are some limitations due to the possibility of matched donors and the risk of graft-versus-host disease (GVHD). Other possibilities are under consideration, like the use of genetic modification (clustered regularly interspaced short palindromic repeats (CRISPR) technology) of patient autologous cells. Also, the use of drugs that promote an increase in HbF, greatly improving the phenotype of patients with β-thalassemia, is under consideration [12, 23].
Other therapeutic proposals, still in the experimental phase, involve the use of inhibitors of JAK2, which is a major player in inducing splenomegaly, frequent in patients with β-thalassemia, and thus avoiding its removal in the most severe forms [12, 14].
Furthermore, NTDT patients, during the progression of the disease, can undergo iron overload caused by an increased absorption, which tends to accumulate and damage different organs. Recently, the clinical use of minihepcidins is under evaluation, or a liver-produced hepcidin analogue able to limit iron overload thus improving the quality of life of β-thalassemia patients [24].
This work aims not only at highlighting the difficulties in establishing a correct diagnosis of β-thalassemia in the cases of an initially nonspecific clinical picture, but also to address problems linked to language barrier, which made communication between health care personnel and the patient particularly difficult. Currently, the language barrier is recognized by health care personnel as one of the main obstacles in providing health care. This problem lacks an appropriate general solution. Often, “rudimentary” solutions are used, such as the use of paper dictionaries or online translators, thus lengthening the timeframe for diagnosis and intervention, and fundamental actions, especially in severe cases. However, from an ethical point of view, the sharing of personal information should be considered. In the clinical case described in this article, an online Arabic translator was used during the preliminary interview, thus allowing the medical staff to obtain the contact of an English-speaking acquaintance who was permitted to know all clinical information about the patient.
In conclusion, the language barrier solution in health care could be done by different ways, for example by providing hospitals with easily accessible telephone translation services, or with the use of artificial intelligence (AI) interpreters in the future. However, these solutions could require the use of a large amount of funds and economic resources that at the moment are not available [25].
Learning points
Considering the possibility to classify patients according to the transfusion needs, our clinical case can be included in the TDT category, independently from the molecular findings that suggest a β-thalassemia intermedia. In fact, several modifier genes and loci associated with clinical phenotype modification have been discovered using new next-generation sequencing (NGS) techniques [26]. These modifier mutations (e.g., BCL11A) can either ameliorate the disease phenotype by increasing the synthesis of HbF [27] or worsen the clinical phenotype by coinheritance of excess of the α-globin gene expression [28]. Other gene mutations or loci, not involved in globin cluster, have been reported to improve the effects on the severity of β-thalassemia. For example, intergenic variants present in a regulatory region between HBS1L and MYB genes contain controlling MYB expression [29]. Coinheritance of these determinants and α-thalassemia ameliorates β-thalassemia [30, 31]. Other comorbidities, like the reaction to transfusion as in this case report, can worsen the degree of the disease. These aspects should be considered and taken into account for a specific individual treatment in the frame of personalized medicine program.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
The authors declare that they have no conflict of interest.
Informed Consent
Informed consent has been obtained, and the study was conducted in accordance with the Declaration of Helsinki.
Author Contributions
AT, SB and GA wrote and revised the paper. MM and MP analyzed the data. GV, CC, and FC performed the analysis. MI, and RM collected the data.
Data Availability
The data supporting this study findings are not publicly available due to the need to protect patient privacy, but are available on reasonable request from the corresponding author, AT.
| References | ▴Top |
- Dell'Edera D, Malvasi A, Tinelli A, Mazzone E, Leo M, Monti V, Epifania AA. [Importance of the molecular diagnosis in the screening of alpha-thalassemia]. Recenti Prog Med. 2011;102(7-8):302-306.
doi pubmed - Muncie HL, Jr., Campbell J. Alpha and beta thalassemia. Am Fam Physician. 2009;80(4):339-344.
pubmed - Harteveld CL, Higgs DR. Alpha-thalassaemia. Orphanet J Rare Dis. 2010;5:13.
doi pubmed pmc - Li CK. New trend in the epidemiology of thalassaemia. Best Pract Res Clin Obstet Gynaecol. 2017;39:16-26.
doi pubmed - Henderson S, Timbs A, McCarthy J, Gallienne A, Van Mourik M, Masters G, May A, et al. Incidence of haemoglobinopathies in various populations - the impact of immigration. Clin Biochem. 2009;42(18):1745-1756.
doi pubmed - Viprakasit V, Ekwattanakit S. Clinical Classification, Screening and Diagnosis for Thalassemia. Hematol Oncol Clin North Am. 2018;32(2):193-211.
doi pubmed - Khan I, Shaikh H. Cooley Anemia. StatPearls Publishing LLC; 2023.
- Sabath DE. Molecular diagnosis of thalassemias and hemoglobinopathies: an ACLPS critical review. Am J Clin Pathol. 2017;148(1):6-15.
doi pubmed - Bennett LF, Liao C, Paulson RF. Stress Erythropoiesis Model Systems. Methods Mol Biol. 2018;1698:91-102.
doi pubmed pmc - Rivella S. Iron metabolism under conditions of ineffective erythropoiesis in beta-thalassemia. Blood. 2019;133(1):51-58.
doi pubmed pmc - Ravasi G, Pelucchi S, Buoli Comani G, Greni F, Mariani R, Pelloni I, Bombelli S, et al. Hepcidin regulation in a mouse model of acute hypoxia. Eur J Haematol. 2018;100(6):636-643.
doi pubmed - Rivella S. beta-thalassemias: paradigmatic diseases for scientific discoveries and development of innovative therapies. Haematologica. 2015;100(4):418-430.
doi pubmed pmc - Kautz L, Jung G, Du X, Gabayan V, Chapman J, Nasoff M, Nemeth E, et al. Erythroferrone contributes to hepcidin suppression and iron overload in a mouse model of beta-thalassemia. Blood. 2015;126(17):2031-2037.
doi pubmed pmc - Libani IV, Guy EC, Melchiori L, Schiro R, Ramos P, Breda L, Scholzen T, et al. Decreased differentiation of erythroid cells exacerbates ineffective erythropoiesis in beta-thalassemia. Blood. 2008;112(3):875-885.
doi pubmed pmc - Weatherall DJ. The definition and epidemiology of non-transfusion-dependent thalassemia. Blood Rev. 2012;26(Suppl 1):S3-6.
doi pubmed - El-Latif MA, Filon D, Rund D, Oppenheim A, Kanaan M. The beta+-IVS-I-6 (T—>C) mutation accounts for half of the thalassemia chromosomes in the Palestinian populations of the mountain regions. Hemoglobin. 2002;26(1):33-40.
doi pubmed - Gibbons RJ, Pellagatti A, Garrick D, Wood WG, Malik N, Ayyub H, Langford C, et al. Identification of acquired somatic mutations in the gene encoding chromatin-remodeling factor ATRX in the alpha-thalassemia myelodysplasia syndrome (ATMDS). Nat Genet. 2003;34(4):446-449.
doi pubmed - Nienhuis AW, Nathan DG. Pathophysiology and Clinical Manifestations of the beta-Thalassemias. Cold Spring Harb Perspect Med. 2012;2(12):a011726.
doi pubmed pmc - Brunner AM, Steensma DP. Myelodysplastic syndrome associated with acquired beta thalassemia: "BTMDS". Am J Hematol. 2016;91(8):E325-327.
doi pubmed pmc - Gologan R. Mixed myelodysplastic syndrome associated with beta-thalassemia intermedia. Leuk Res. 2010;34(8):e221-223.
doi pubmed - Filon D, Oron V, Krichevski S, Shaag A, Shaag Y, Warren TC, Goldfarb A, et al. Diversity of beta-globin mutations in Israeli ethnic groups reflects recent historic events. Am J Hum Genet. 1994;54(5):836-843.
pubmed pmc - Bahrani S, Teimouri-Jervekani Z, Sadeghi M. Thrombotic events and anticoagulants in beta-thalassemia patients with focus on anticoagulants for atrial fibrillation: a brief review. Curr Probl Cardiol. 2022;47(9):100912.
doi pubmed - Dong A, Rivella S, Breda L. Gene therapy for hemoglobinopathies: progress and challenges. Transl Res. 2013;161(4):293-306.
doi pubmed pmc - Preza GC, Ruchala P, Pinon R, Ramos E, Qiao B, Peralta MA, Sharma S, et al. Minihepcidins are rationally designed small peptides that mimic hepcidin activity in mice and may be useful for the treatment of iron overload. J Clin Invest. 2011;121(12):4880-4888.
doi pubmed pmc - Squires A. Strategies for overcoming language barriers in healthcare. Nurs Manage. 2018;49(4):20-27.
doi pubmed pmc - https://www.ithanet.eu/db/ithagenes?action=stats.
- Tripathi P, Agarwal S, Mandal K, Gupta A, Sarangi AN. Impact of genetic polymorphisms in modifier genes in determining fetal hemoglobin levels in beta-thalassemia. Thalassemia Reports. 2023;13(1):85-112.
- Habara A, Steinberg MH. Minireview: Genetic basis of heterogeneity and severity in sickle cell disease. Exp Biol Med (Maywood). 2016;241(7):689-696.
doi pubmed pmc - Langer AL. Beta-Thalassemia. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH et al., editors. GeneReviews(R). Seattle (WA). 2023.
- Galanello R, Sanna S, Perseu L, Sollaino MC, Satta S, Lai ME, Barella S, et al. Amelioration of Sardinian beta0 thalassemia by genetic modifiers. Blood. 2009;114(18):3935-3937.
doi pubmed pmc - Minaidou A, Stephanou C, Tamana S, Xenophontos M, Lederer C, Kountouris P, et al. P128: Ithanet: an information and database community portal for haemoglobinopathies. Hemasphere. 2022;6(Suppl):31.
doi
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.