

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://www.thejh.org |
Case Report
Volume 13, Number 4, August 2024, pages 174-177

Is There a Correlation Between Immune Thrombocytopenia and Immunoglobulin G4-Related Disease?
Dorela Lamea, Michelangelo Pianellia, Erika Morsiaa, Attilio Olivieria, Antonella Polonia, b
aUniversita Politecnica delle Marche, AOU delle Marche, Ancona, Italy
bCorresponding Author: Antonella Poloni, Dpt Scienze Cliniche e Molecolari, Universita Politecnica delle Marche, AOU delle Marche, 60126 Ancona, Italy
Manuscript submitted March 11, 2024, accepted May 15, 2024, published online August 15, 2024
Short title: ITP and IgG4-RD
doi: https://doi.org/10.14740/jh1260
| Abstract | ▴Top |
Immunoglobulin G4-related disease (IgG4-RD) is an immune-mediated condition causing organ swelling and fibrosis. Rarely, it coexists with primary immune thrombocytopenia (ITP), characterized by low platelet count (< 100 × 106/L) without an underlying cause. We present a case of a 56-year-old woman diagnosed with ITP in 2005, successfully treated with dexamethasone and intravenous immunoglobulins (IVIG). In 2011, she was diagnosed with IgG4-RD, type I autoimmune pancreatitis, initially treated with steroids then azathioprine with no response. ITP relapses were managed with prednisone/IVIG, rituximab, and thrombopoietin-receptor agonist therapy. Fostamatinib provided temporary relief, but platelet count dropped again in 2023. Combination therapy with small doses of prednisone and mycophenolate showed a partial response, maintaining platelet count over 50 × 106/L. Further investigation is warranted to explore any correlation between these two conditions, especially considering the patient’s prolonged response to immunosuppressors.
Keywords: ITP; IGG4-RD; TPO-RA; Immunosuppressor
| Introduction | ▴Top |
Immune thrombocytopenia (ITP) is a complex autoimmune disease characterized by reduced platelet count resulting from both platelet destruction and impaired platelet production. Current estimates suggest an incidence of 1.9 to 3.9 per 100,000 individuals [1]. The disorder, characterized by its heterogeneity, presents with a range of clinical symptoms. Bleeding events in ITP patients tend to be unpredictable. Even in the presence of severe thrombocytopenia, manifestations may be limited to bruising and petechiae. However, more severe mucosal bleeding, such as menorrhagia, epistaxis, gastrointestinal hemorrhage, hematuria, or, rarely, intracranial hemorrhage (ICH), may occur. Diagnosis of ITP typically involves the exclusion of other causes of thrombocytopenia [1].
The clinical course of ITP varies based on its classification. This includes primary ITP (not associated with other conditions), occurrence alongside additional autoimmune cytopenias (Evans syndrome), manifestation as part of a primary immunodeficiency, or connection to an underlying autoimmune condition or infection (secondary ITP) [2].
While secondary ITP may co-occur with other autoimmune disorders, the precise mechanisms underlying this association remain unknown. In the pathogenesis of ITP, it is hypothesized that both B cell-mediated and T cell-mediated mechanisms play integral roles [3].
Here we describe the case of one of our patients diagnosed with ITP and concomitantly diagnosed with an immunoglobulin G4-related disease (IgG4-RD). Clinically, this IgG4-RD is characterized by autoimmune pancreatitis of type 1 [4, 5].
| Case Report | ▴Top |
Herein, we present a case report of a 56-year-old woman admitted to our clinic in May 2005 for severe thrombocytopenia (platelets 1 × 106/L) and a petechial rash on the lower limbs. In the blood tests at onset, platelets were not measurable. Hemoglobin levels were measured at 11.4 g/dL, white blood cell count at 4.6 × 106/L, and coagulation markers fell within the normal range. The peripheral blood smear displayed no schistocytes, no features of hematologic disorders, and no visible platelets. Additionally, the serum protein electrophoresis and serum electrolytes, lactate dehydrogenase, D-dimer levels, renal function and liver function all yielded normal range. Further investigations included negative serology for hepatitis C virus (HCV), human immunodeficiency virus (HIV), hepatitis B virus (HBV), parvovirus, negative Epstein-Barr virus (EBV)-DNA and cytomegalovirus (CMV)-DNA, and a negative Helicobacter pylori (H. pylori) antigen test in feces. The antinuclear antibodies (ANA), extractable nuclear antigens (ENA), antineutrophil cytoplasmic antibodies (ANCA), and anti-phospholipid antibodies all fell within their respective normal ranges. The only detected anomaly was a positive result in both direct and indirect Coombs tests, in the absence of anemia or hemolysis. Both a cerebral computed tomography (CT) scan and chest radiography yielded negative results. A bone marrow aspirate and biopsy revealed small megakaryocytes not clustered and a reactive infiltration, excluding hematologic disorders. Given the lack of definitive findings from the screening tests, bone marrow evaluation and the unresponsiveness to platelet transfusions, a presumptive treatment for ITP was initiated. This treatment included dexamethasone at a dose of 40 mg for 4 days and human intravenous immunoglobulins (IVIG) at a dosage of 2 g/kg (1 g/kg for 2 days) resulting in complete platelet recovery (Fig. 1). The patient has been enrolled in a follow-up program.
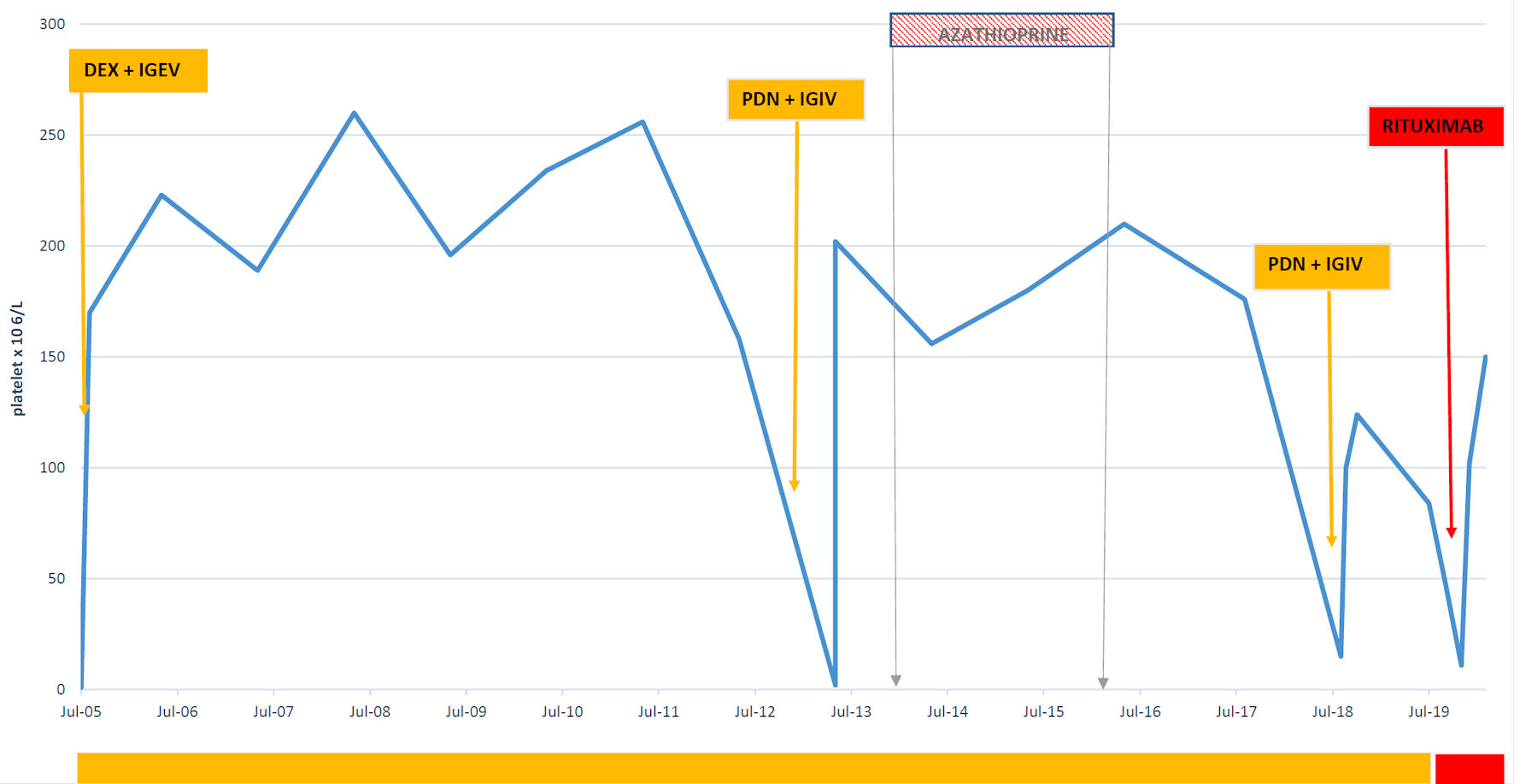 Click for large image | Figure 1. Platelet variation from 2005 till July 2019. Dex: dexamethasone; IVIG: intravenous immunoglobulin; PDN: prednisone. |
In 2011, during a follow-up appointment, the patient reported experiencing abdominal pain. Laboratory examinations revealed significantly elevated levels of amylases and lipases. Subsequent abdominal ultrasonography identified an enlarged pancreas, particularly in the transition between the body and isthmus, and the body and tail and a mesenteric mass measuring approximately 10 cm.
The initial hypothesis was pancreatic neoplasia, prompting the patient to undergo an exploratory laparotomy confirming the abdominal mass and requiring en bloc jejunoileal resection coupled with an ileo-jejunal anastomosis. Biopsies were performed on the pancreatic lesions. Both the removed mass and the pancreatic biopsies were evaluated extemporaneously showing no evidence of any tumor but rather infiltration of lymphoplasmacytic elements, leading to the decision not to proceed with a pancreatectomy. Later on, histology indicated an IgG4-positive inflammatory pseudotumor with serum IgG4 > 250 mg/mL, concluding for an IgG4-RD like chronic autoimmune pancreatitis type 1 (AIP1). Initial steroid therapy elicited a prompt serum IgG4 response, but little response in the pseudotumor reduction.
So due to pseudotumor persistence on an abdomen CT scan in 2014, along with the persistence of abdominal pain and worsening diabetes, requiring occasional insulin therapy, it was decided to initiate a second-line therapy with azathioprine. On a routine abdomen CT in 2016, the pancreatic mass in the transition between the body and tail was stable, so azathioprine was interrupted. The patient underwent an ultrasound-guided biopsy that confirmed the diagnosis of AIP1.
In the follow-up period, in 2013, the first relapse of ITP occurred, characterized by gingival bleeding, widespread hematomas, and a petechial rash. The prescribed treatment involved administering prednisone at a dosage of 1 mg/kg, along with IVIG. Subsequently, a low-dose maintenance therapy with prednisone was implemented, yielding an outstanding response in terms of platelet count.
The second relapse occurred in 2018, presenting with minor hemorrhagic symptoms and a platelet count of 3 × 106/L. Another course of steroids achieved a complete response that endured for nearly a year. However, the third relapse in 2019, characterized by a platelet count of 8 × 106/L and the onset of type II insulin-dependent diabetes, prompted the administration of rituximab. This resulted in complete remission, which persisted for another year.
The fourth recurrence in November 2020 required treatment with TPO mimetic; initially eltrombopag, followed by romiplostim from November 2021 to January 2022. Unfortunately, subsequent blood examinations revealed a platelet count of 0, leading to the discontinuation of romiplostim. Fostamatinib was initiated, resulting in complete remission without the need for steroid maintenance. A 2022 abdomen CT scan revealed a persistent pancreatic lesion, albeit stable compared to the previous scan.
From February 2023, a gradual reduction in platelet counts ensued, reaching 8 × 106/L in April. The reintroduction of a low dose of steroids elicited minimal response. In collaboration with a rheumatologist, a decision was made to explore immunomodulatory therapy with mycophenolate and steroid maintenance, leading to a good response in the platelet count. As of February 2024, the patient is currently undergoing treatment with mycophenolate 1,500 mg and prednisone 2.5 mg on alternate days. The platelet count stands at 64 × 106/L. In November 2023, the patient also experienced a coronavirus disease 2019 (COVID-19) infection without any fluctuations in the platelet count (Fig. 2).
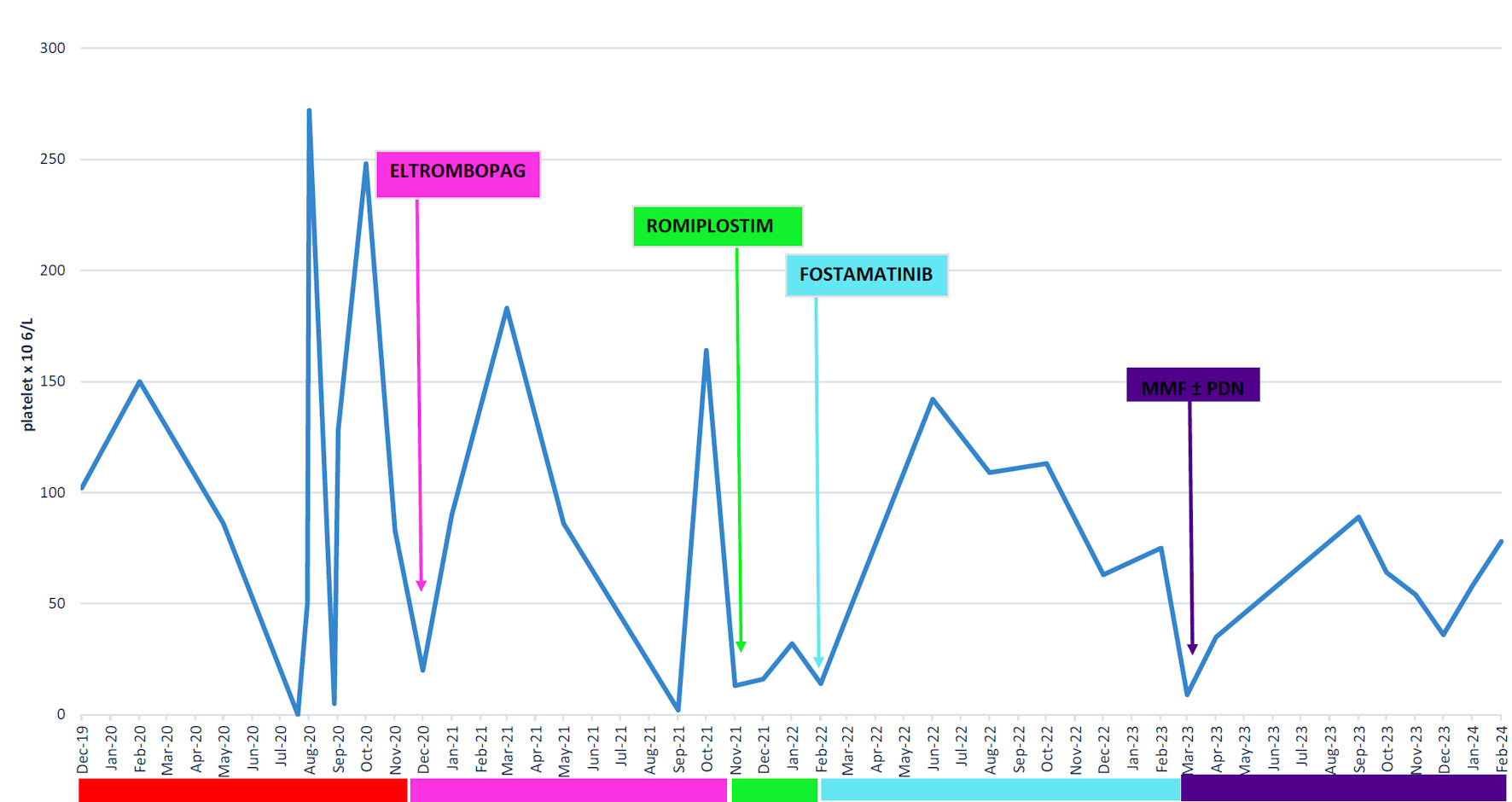 Click for large image | Figure 2. Platelet variation from July 2019 till February 2024. MMF: mycophenolate; PDN: prednisone. |
| Discussion | ▴Top |
ITP represents a prevalent thrombocytopenic disorder, and the comprehension of its pathophysiology is currently in its nascent stages. Emerging insights suggest that B- and T-cell defects play a pivotal role in the pathophysiology of ITP, with compelling evidence indicating that platelet autoimmunity, primarily involving IgG, arises from a breakdown in self-tolerance mechanisms [3]. Concerning IgG4-RD, it represents an immune-mediated chronic fibro-inflammatory disorder typified by tumefactive lesions, dense lymphoplasmacytic infiltrates, and an abundance of IgG4-bearing plasma cells within the affected tissue [6]. IgG4 plays a role in autoimmune diseases other than rheumatic diseases, such as pemphigus, bullous pemphigoid, idiopathic membranous glomerulonephritis, or myasthenia gravis, but also in helminth infections. Research shows the importance of IgG4 in malignancy of neoplasms. Melanoma cells are known to stimulate IgG4 production through a modified Th2-based inflammatory response [7].
While our patient received a diagnosis of ITP prior to the diagnosis of AIP1, it remains unclear whether the latter disease manifested itself only when the mass was substantial and absent before the ITP diagnosis. This ambiguity arises, in part, because high doses of cortisone, also used in the treatment of AIP1, were necessary both at the diagnosis of ITP and during the initial relapse. It is crucial to ascertain whether there might be a correlation in this patient between the two conditions, specifically, whether the IgG4 plasma cells associated with AIP1 could produce IgG4 antibodies against platelets, leading to their destruction, or if circulating IgG4 antibodies could activate T cell-mediated platelet destruction.
In an attempt to address these questions, we conducted a literature review and identified a study by Chan et al that delved into the IgG subclass distribution of IgG anti-GPIIb/IIIa autoantibodies bound to platelets in ITP patients [8]. The objective was to discern whether the findings could substantiate either a polyclonal or an oligoclonal model of autoantibody production. The study revealed that 30% of patients exclusively exhibited IgG1 autoantibodies, while an additional 43% presented with IgG1 accompanied by other IgG subclass antibodies. Intriguingly, certain ITP patients exhibited a notable subclass restriction, including 7% confinement to subclass 4. This outcome was unexpected, considering the well-known low binding affinity of IgG4 with the Fc receptor of phagocytic cells, contrasting with reported subclass frequencies from other studies [9, 10]. Nevertheless, research has indicated no significant difference in the ability of different IgG subclasses to mediate T-cell activation [11].
It should be noted that our patient exhibited positive responses predominantly to immunosuppressants such as steroids, rituximab, and mycophenolate. Given these observations, we find it pertinent to explore the potential role of serum IgG4 in the pathophysiology of ITP.
Conclusions
ITP is a heterogeneous disease that can manifest either as a primary or secondary condition. Understanding the physiopathological differences between these two categories is crucial. It would be beneficial to explore how autoimmune diseases, such as IgG4-RD, can influence the clinical presentation, course, and the most suitable therapeutic approaches.
Acknowledgments
We would like to thank the patient and their family for their cooperation, as well as the medical staff, colleagues, and our institution for their invaluable support and contributions to this case report.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
The study was conducted in accordance with Good Clinical Practice guidelines and the Declaration of Helsinki and was approved by “Azienda Ospedaliera Universitaria delle Marche” Ethics Committee. The patient has given consent and has been enrolled in the clinical protocol NCT03465020.
Author Contributions
Conceptualization: AP. Writing, review and editing: DL, MP, AP. Editing: EM, AO. All authors have read and agreed to the published version of the manuscript.
Data Availability
Any inquiries regarding supporting data availability of this study should be directed to the corresponding author.
| References | ▴Top |
- Kohli R, Chaturvedi S. Epidemiology and clinical manifestations of immune thrombocytopenia. Hamostaseologie. 2019;39(3):238-249.
doi pubmed - Provan D, Arnold DM, Bussel JB, Chong BH, Cooper N, Gernsheimer T, Ghanima W, et al. Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood Adv. 2019;3(22):3780-3817.
doi pubmed pmc - Provan D, Semple JW. Recent advances in the mechanisms and treatment of immune thrombocytopenia. EBioMedicine. 2022;76:103820.
doi pubmed pmc - Lohr JM, Beuers U, Vujasinovic M, Alvaro D, Frokjaer JB, Buttgereit F, Capurso G, et al. European Guideline on IgG4-related digestive disease - UEG and SGF evidence-based recommendations. United European Gastroenterol J. 2020;8(6):637-666.
doi pubmed pmc - Guma M, Firestein GS. IgG4-related diseases. Best Pract Res Clin Rheumatol. 2012;26(4):425-438.
doi pubmed - Tarte NN, Ravipati CS, Leon de la Rocha JA, Rinker E, Patel NJ. IgG4-related disease with multiorgan involvement: a case-based review. Rheumatol Int. 2021;41(6):1169-1174.
doi pubmed - Maslinska M, Dmowska-Chalaba J, Jakubaszek M. The Role of IgG4 in autoimmunity and rheumatic diseases. Front Immunol. 2021;12:787422.
doi pubmed pmc - Chan H, Moore JC, Finch CN, Warkentin TE, Kelton JG. The IgG subclasses of platelet-associated autoantibodies directed against platelet glycoproteins IIb/IIIa in patients with idiopathic thrombocytopenic purpura. Br J Haematol. 2003;122(5):818-824.
doi pubmed - Tijhuis GJ, Klaassen RJ, Modderman PW, Ouwehand WH, von dem Borne AE. Quantification of platelet-bound immunoglobulins of different class and subclass using radiolabelled monoclonal antibodies: assay conditions and clinical application. Br J Haematol. 1991;77(1):93-101.
doi pubmed - Aster RH, George JN, McMillan R, Ganguly P. Workshop on autoimmune (idiopathic) thrombocytopenic purpura: pathogenesis and new approaches to therapy. Am J Hematol. 1998;58(3):231-234.
doi pubmed - Jelley-Gibbs DM, Plitnick LM, Gosselin EJ. Differences in IgG subclass do not effect immune complex-enhanced T cell activation despite differential binding to antigen presenting cells. Hum Immunol. 1999;60(6):469-478.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.