

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://www.thejh.org |
Review
Volume 13, Number 3, June 2024, pages 53-60

Sickle Cell Screening in Adults: A Current Review of Point-of-Care Testing
Sebastian R. Mendez-Martia, c, Chad Zika, Sheinei Alana, Hongkun Wangb, William B. Ershlera
aAdult Sickle Cell Center, Inova Schar Cancer Institute, Inova Fairfax Medical Campus, Fairfax, VA, USA
bBiostatics, Bioinformatics and Biomathematics Department, Georgetown University, Washington, DC, USA
cCorresponding Author: Sebastian R. Mendez-Marti, Adult Sickle Cell Center, Inova Schar Cancer Institute, Fairfax, VA 22031, USA
Manuscript submitted April 3, 2024, accepted June 10, 2024, published online June 28, 2024
Short title: Review of Sickle Cell Screening Tests
doi: https://doi.org/10.14740/jh1272
- Abstract
- Sickle Cell Screening With the Sickle Cell Solubility Test (SCST)
- SCT
- Historical and Current Applications of the SCST
- Review of Evidence Supporting Screening Protocols
- Community Awareness of Sickle Cell Screening and the SCST
- Future Directions Beyond the Solubility Test
- References
| Abstract | ▴Top |
In adults, the sickle cell solubility test (SCST) is the most common screening test to determine the presence of hemoglobin S (HbS) within a blood sample. The assay is inexpensive, rapid, highly sensitive and specific. However, the SCST cannot accurately quantify the level of HbS in a test sample and requires confirmatory testing to distinguish between sickle trait and sickle cell disease. Despite these limitations, it remains the standard screening tool for HbS in a variety of settings such as screening in the US military or by the National Collegiate Athletic Association. With an increased awareness of the importance of screening for sickle cell in adults, we herein describe the current sensitivity, specificity, positive predictive value, and negative predictive value of this test. We also review overall clinical utility of this laboratory measure and briefly discuss new point-of-care techniques designed to overcome the SCST’s shortcomings.
Keywords: Sickle cell solubility test; Sickle cell screening; Point-of-care screening; Sickle cell disease; Sickle cell trait
| Sickle Cell Screening With the Sickle Cell Solubility Test (SCST) | ▴Top |
The ability to screen patients for sickle cell disease (SCD) was established with the development of the SCST. The SCST, commonly known as “sickle prep” is a qualitative assay used to detect the presence of hemoglobin S (HbS) within a blood sample. The SCST was discovered in 1949 following the discovery that sodium dithionate, a reducing agent, induced sickling of red blood cells by deoxygenation of HbS [1, 2]. Subsequently, this technology was refined in the 1970s when Ortho Diagnostics developed an assay under the trade name Sickledex® analogous to the modern “sickle prep” [2, 3]. The SCST was standardized following the formation of the Hemoglobinopathy Reference Laboratory at the Centers for Disease Control and Prevention (CDC) in 1972, which published procedural manuals for laboratories and offered training courses to include the SCST until 1993 when these services were transitioned to the Newborn Screening Quality Assurance Laboratory [2].
The SCST relies on the property that HbS is insoluble and precipitates in solution when mixed with a reducing agent in a phosphate buffer [3, 4]. The most common reducing agent used to precipitate the solution is sodium hydrosulfite, also known as sodium dithionite [5-8]. The test is performed by adding 0.02 mL of whole blood to 2 mL of a solution of sodium hydrosulfite. After remaining for 2 - 5 min at room temperature, the tubes are examined against a printed background. Blood samples drawn from individuals with circulating HbS appear cloudy (Fig. 1) indicating a positive test [3, 7]. A negative test is shown by a transparent solution.
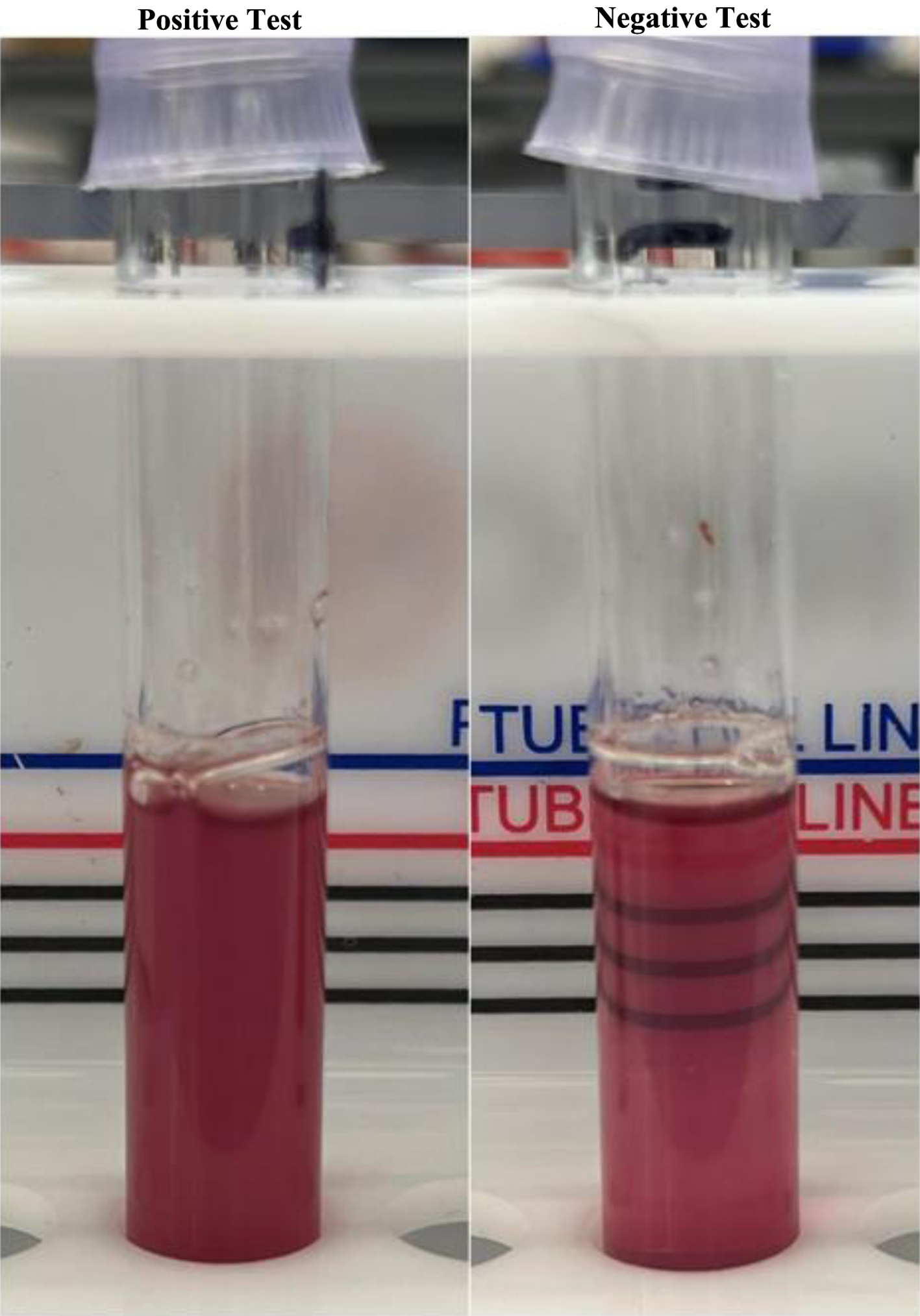 Click for large image | Figure 1. Sickle cell solubility test in the laboratory. The presence of hemoglobin S is marked by a turbid solution (on the left). The control solution is clear, allowing for visualization of the lines (right). |
The SCST has been reported to be highly sensitive (98.9%) and specific (100%) for detection of HbS [6]. Conversely, in an African study of 200 patients between the age of 6 months and 5 years, the SCST was reported to be 45% sensitive (95% confidence interval (CI): 25.8 - 65.8) and 90% specific (95% CI: 84.8 - 93.6) when compared with concurrent hemoglobin electrophoresis (HE) [9]. The false positive rate was attributed to erythrocytosis, leukocytosis or hyperlipidemia but this could not be confirmed. False negatives have been reported in patients with severe anemia or high levels of fetal hemoglobin (HbF) [5, 6]. We hypothesize this occurs at low HbS concentration such that it is below the detection limit of the assay.
We investigated the sensitivity, specificity, positive predictive value (PPV) and negative predictive values (NPV) of the SCST through retrospective chart review in adult patients seen at our institution by one of our center providers. During the initial study period (July 2023), we examined the two most recent SCSTs compared to concomitantly drawn HE in 112 patients with known SCD. This approach yielded 200 distinct SCST results with associated HbS levels. Twenty-four (24) patients only had one SCST available to be analyzed. After our primary analysis, we also examined the two most recent SCSTs compared to concomitantly drawn HE in 16 patients with sickle cell trait (SCT) and one patient without SCD or SCT (AA genotype). SCT patients who were positive for the SCST were considered a false positive for the SCST. Four (4) patients had two SCSTs with concomitantly drawn HE, allowing for 20 samples to be examined. Combined, this yielded a total of 220 SCST samples to be analyzed. Twenty-five individuals (or 22% of the 112 SCD patients) underwent red cell exchange during this period. Patient information was de-identified after the initial data collection.
Six (6) of the 220 samples were negative for the SCST (Fig. 2). Four (4) out of the six patients were SCD patients who had undergone red cell exchange, one patient was heavily transfused prior to SCST collection, and the last was the hemoglobin AA patient. Using the presence of SCD as the true disease status, the overall sensitivity and specificity of the SCST was 97.5% and 5%, respectively. The PPV was 91% and the NPV was 17% based on the prevalence in this clinic population. Two hundred seven samples had HbS levels 15% or above, and these were all positive for the SCST. However, all six samples that were negative by the SCST were included in the 13 samples that had HbS levels below 15%. When correcting for HbS concentration, calculated as the HbS% multiplied by the total hemoglobin from the complete blood count, the limit of detection appears to be approximately 1.2 g/L HbS. Based on our analysis, the SCST is sensitive to detect HbS in patients with SCD, with limitations once the HbS levels are less than 15% or the HbS concentration is less than 1.2 g /L HbS.
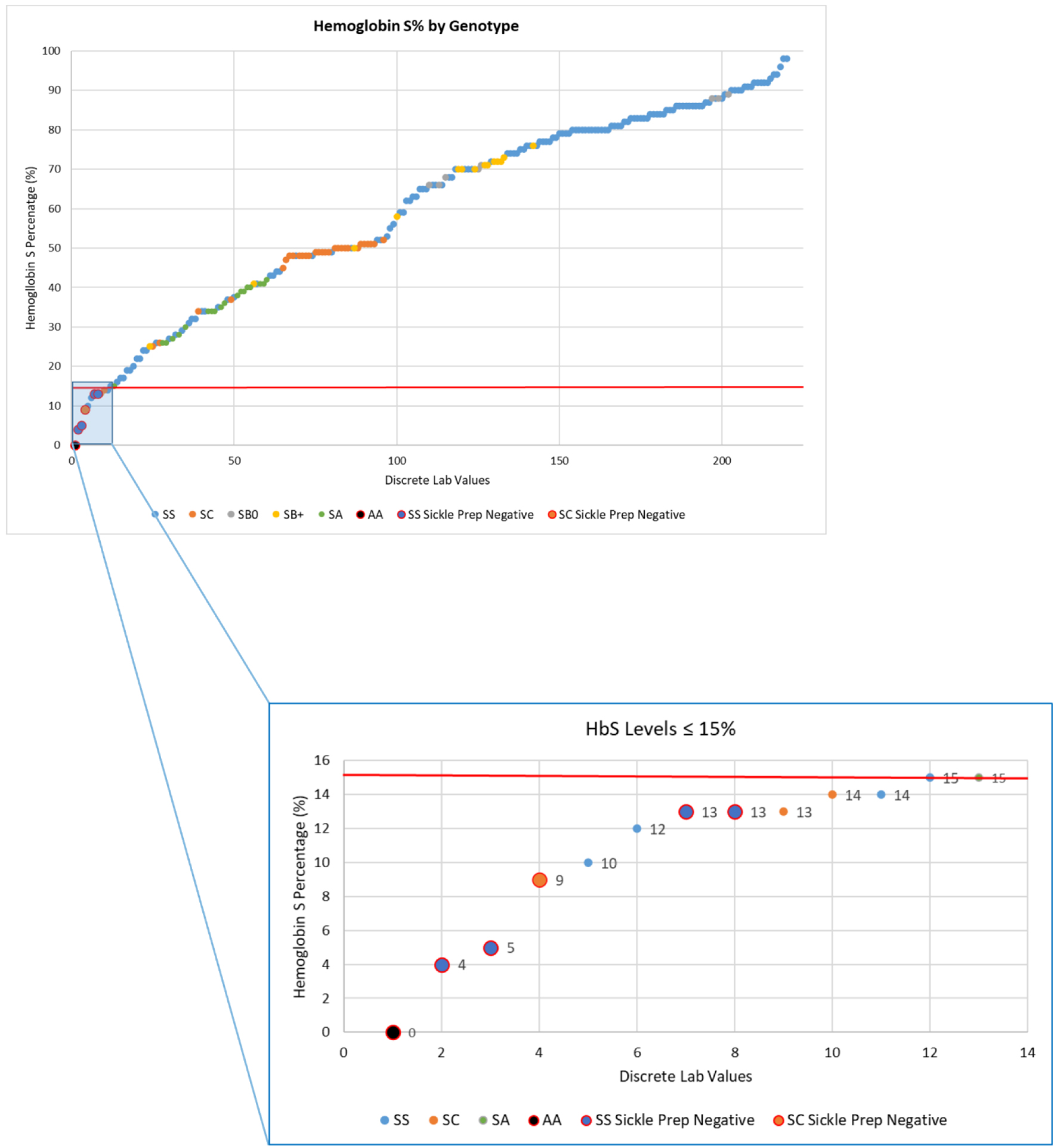 Click for large image | Figure 2. Hemoglobin S (HbS) percentage with concomitant sickle cell solubility testing (SCST) in 220 distinct samples in patients with sickle cell disease (SCD), sickle cell trait (SCT) or HbAA. These samples were taken from 112 patients with SCD of all genotypes, 16 patients with SCT, and one patient without SCD or SCT. SCD patients were further subdivided by genotype: HbSS, HbSC, HbS-beta thalassemia null, or HbS-beta thalassemia positive. Of the 220 samples, five were false negative (large circles). These all occurred at an HbS level < 15%. This subset is highlighted in the blue subsection with individual negative values indicated by the large circles. |
As the SCST is a qualitative test, it cannot distinguish between SCT and sickle cell anemia, nor does it quantify the degree of HbS. Given these limitations, the sickle solubility test is not used for newborn screening programs as the dilutional effect of HbF can persist up to 6 months and this would predictably be associated with false negative results [5, 10, 11]. Newborn screening in the United States began after the multicenter Cooperative Study of Sickle Cell Disease (CSSCD) identified that children with SCD had a 20% case fatality rate from pneumococcal sepsis, and that daily prophylactic dosing of penicillin was associated with 84% reduction in incidence of infection in the subsequent Prophylactic Penicillin Study published in 1986 [2, 12]. This findings prompted the National Heart, Lung and Blood Institute (NHLBI) at the National Institutes of Health (NIH) to recommend universal newborn screening in 1987 as part of the “Newborn Screening for Sickle Cell Disease and Other Hemoglobinopathies” Conference [2, 13]. Despite increased available funding, the uptick of universal newborn screening was slow and did not become standard procedure across the United States until 2007 [2, 11]. Newborn screening utilizes more labor-intensive procedures such as isoelectric focusing and high-performance liquid chromatography [11]. In the last decade, there has been renewed interest in adult screening following reports of adverse outcomes with adults with SCT and increased awareness of SCD in adults [14]. As screening in adults is primarily conducted using the SCST, this review will subsequently discuss SCT, the application and methodology for screening in US military and college athletes followed by recent technological advances regarding sickle cell point-of-care testing.
| SCT | ▴Top |
SCT is present in one out 12 African Americans and up to 1.5% of neonates born in the United States [15-17]. Worldwide, the prevalence of SCT is estimated to exceed 300 million individuals primarily in Africa and Southern Asia which are affected by malaria [17, 18]. SCT emerged as a favorable evolutionary advantage in Africa due to increased protection against severe malaria [18].
Although SCT is commonly thought of as a benign condition with normal life expectancy, there is emerging literature suggesting an increased incidence of potentially preventable adverse outcomes in several organ systems [14, 19, 20]. This has been attributed to sickling of HbS in patients with SCT particularly under increased physiologic stress including severe tissue hypoxia, acidosis, increased viscosity, dehydration, or hypothermia [11, 20, 21]. HbS levels in people with SCT can vary from 20% to 40% based in part on the number of alpha-globin genes expressed [14, 19].
Acknowledged end-organ consequences of SCT are variable. From a renal perspective, individuals with SCT are less able to concentrate urine compared to patients without SCT (hyposthenuria), more likely to develop hematuria, and are at increased risk of developing acute kidney injury and chronic kidney disease compared to the general population [19, 21, 22]. From a vascular perspective, individuals with SCT are more at risk for venous thromboembolism, particularly pulmonary emboli relative to deep vein thromboses [14, 17]. SCT patients can also develop splenic infarcts [14, 19]. The association of SCT with pregnancy-related outcomes remains unclear [14, 17]. SCT is also associated with development of renal medullary carcinoma. Based on a literature review of 166 patients from 47 studies, 98% of cases occurred in individuals with SCT [23]. In addition to increased surveillance of complications of SCT, an understanding of the heritable characteristics of SCD may prove very useful for those with trait. There is a growing effort to screen at risk adults and make accessible education and preventive measure for those who test positively.
| Historical and Current Applications of the SCST | ▴Top |
Screening for sickle cell anemia and SCT gained national attention in the United States following publication of several case reports highlighting catastrophic occurrences of exertional related illness among otherwise healthy individuals with SCT [24]. After a 1981 investigation of SCT and exertional heat related injuries, the Department of Defense adopted a policy for universal screening of all military recruits utilizing the SCST [25]. Positive results were followed by HE to confirm and quantify the presence of HbS (either SS or SA) with a cutoff of HbS < 41% to qualify for participation of high risk activities [25]. The policy was withdrawn in 1985 after concerns for stigma, and reinstated following the three US Air Force Recruit deaths in the 1990s [17, 25].
As a response to these events, in the early 2000s, the US Army instituted a protocol of universal training precautions (UTPs) to decrease exercise-related injury among all recruits irrespective of SCT status. UTPs are universal procedures designed to prevent complications of physical training programs to include bodily injury, illness, and death [26]. Although no formal guideline exists to address what strategies should be implemented to prevent exercise-related health risks, individual organizations have implemented protocols that address advanced assessment of climate factors, adjustment of exertion, consistent hydration, and dedicated recovery time as a prevention strategy for all exertion-related injury [26, 27]. In light of increased awareness of SCD and SCT, the US army screens all recruits for hemoglobinopathies since 2020, and does not allow personnel to enroll who have an HbS > 45% [25, 28]. Practices differ among the different branches of the US military in how recruits with SCT are identified.
The National Collegiate Athletic Association (NCAA) began screening athletes for sickle cell in Division I sports following the sudden death of several athletes and subsequent lawsuit in 2009 [29]. The NCAA implemented mandatory screening for Division I athletes in 2010, which ultimately expanded to Division II and III [29-31]. Student athletes were allowed to opt out of testing but in the most recent iteration of NCAA policy (August 1, 2022 for Division I, II, and III) the opt-out choice was removed [32-34]. Prior to this removal, adherence was heterogeneous as allowance of a waiver was institution dependent [35]. Screening for college athletes is primarily done with the SCST [30, 36].
Outside of military and college athlete screening, the SCST is useful for rapid assessment of the presence of HbS. The British Society of Hematology states it is sensible to utilize the test in the emergency department or prior to anesthesia as a negative test will indicate that the concentration of HbS is low enough as to not increase intraoperative risk [37]. This is followed by the caveat that both positive or equivocal results should be followed by confirmatory testing. The SCST is also used for identification of HbS prior to donation of blood at our own institution and in African Americans wanting to give blood to the American Red Cross [38]. SCT is not a prohibition for donation, and in our institution only patients with SCD are not transfused blood with HbS. However, HbS interferes with the leukoreduction filters and has been associated with prolonged or incomplete filtration of leukocytes [39, 40].
| Review of Evidence Supporting Screening Protocols | ▴Top |
The relationship between SCT and exertional injury is controversial, as the correlation was established in case reports that linked SCT in military recruits to increased risk of exertional rhabdomyolysis, compartment syndrome, splenic infarcts and hyposthenuria [25]. Although retrospective data of military recruits demonstrated an association between SCT and increased rate of morbidity including heat related illness, more recent cohort studies following the implementation of UTP no longer identified this association [25, 41]. A retrospective cohort study conducted using the Stanford Military Data Repository examined the relationship between SCT and rate of exertional rhabdomyolysis and death excluding traumatic injuries from January 2011 to December 2014 in 47,944 Black US Army soldiers, of which 7.4% had SCT [42]. SCT was associated with a higher hazard ratio (HR) for exertional rhabdomyolysis as compared to soldiers without SCT, but the effect was in the same order of magnitude for smokers when compared to non-smokers (HR: 1.54) [42]. Soldiers who recently took statins (HR: 2.89) and antipsychotics (HR: 3.02) demonstrated a greater HR for developing exertional rhabdomyolysis when compared to soldiers who did not use these medications [42]. There was no significant difference in the risk of death among soldiers with SCT as compared with those without SCT (HR: 0.99; 95% CI: 0.46 - 2.13) [42]. In a follow-up study of heat related illness and heat stroke in the above-mentioned population, there was no significant association between heat stroke, mild heat injury and SCT [43]. These findings challenge the assertion that SCT is associated with increased risk of exertional related illness and death, and according to expert opinion, reflects the benefits of UTP [44].
As with the military, the data that support universal testing of student athletes are contentious [45]. A retrospective trial estimated that there were 2,147 Division I athletes with SCT out of 144,181 based on population studies during the 2007 - 2008 academic year [30]. They estimated without intervention about seven student-athletes with SCT would suffer exercise-related sudden death in 10 years [30]. Similarly, examining data from the United States Sudden Death in Athletes Registry over a 31-year period (1980 - 2010), of the 2,462 sudden deaths including survivors of sudden cardiac death, 23 or 0.9% of deaths occurred in athletes with documented SCT based on testing during life or at autopsy [46]. The overall prevalence of SCT in the registry was 2.3% or 699 black athletes [46]. Of the 23 players who had SCT and died, 19 or 83% were football players [46]. When examining the circumstances surrounding the death, all occurred during maximum intensity of exercise, 87% (or 20/23 cases) of the cases occurred under circumstances of high temperatures, and 9% (or 2/23 cases) occurred at high altitude [46]. There was no significant difference between SCT athletes and non-SCT athletes in terms of controlling for other causes of cardiovascular death or age, but this was powered to include African American athletes [46]. In the same registry but looking at sudden deaths of all causes from 1980 to 2006, 1,866 athletes died suddenly [47]. Of these deaths, the most common causes of death were cardiovascular disease to include hypertrophic cardiomyopathy and congenital coronary artery anomalies, accounting for 1,049 deaths or 56% of the studied population [47]. In a separate review of 16 deaths from 2000 to 2010 in Division I college football, 63% (or 10 out of 16 athletes) had SCT [48]. These events were also triggered by several minutes of maximal intensity [48]. The American Society of Hematology in their policy statement recommended against universal screening for athletes as UTPs are effective in reducing exertional related injuries [49]. They also recommended to not mandate testing or disclosure of SCT status as a prerequisite for participation in athletic activities [49]. This approach was supported by the Sickle Cell Disease Association of America and the Advisory Committee on Heritable Disorders in Newborns and Children [50].
| Community Awareness of Sickle Cell Screening and the SCST | ▴Top |
Although newborn screening began in the United States 1972 following the passage of the National Sickle Cell Anemia Control Act and crystallized into national policy in 1987, universal screening was not implemented in all 50 states and the District of Columbia until 2007 [2]. As enactment of the law is state-dependent, there are variations in state procedures on who is notified of a positive result as well as the procedures used to inform patients and medical providers [51]. In a survey-based analysis that assessed each state’s procedures for sickle cell screening, although 100% of patients’ primary care providers (PCPs) were notified of an SCD diagnosis, only 75% of hematologists and 40% of families were also notified [51]. Notification of an SCT diagnosis is lower for families although remains high among PCPs (88% of PCP and 37% of families) [51]. As these results were gleaned from surveys, these are subject to recall bias and the analysis does not specify how the hematologist or primary care doctors were involved in their care.
Consequently, patient’s awareness of their sickle cell status is also low. In a community-based survey of 316 individuals in Northern California in 2006, only 16% knew their trait status [52]. Similarly, in a university-based survey of 258 African American university students, 52% were unsure of their status and 62% did not know if they had family members with SCD [53]. Of the participants who did not know their status, 56% (n = 144) wanted to know their trait status [53].
Although most community members are aware of SCD [52, 54], there is misinformation regarding SCT which may account for the hesitancy in knowing their status. In the above-mentioned California-based survey, although 86.2% (n = 243) of responders were able to identify that SCD causes serious problems, and 70% (n = 197) were able to identify that SCT can cause health problems, 31% incorrectly believed that SCT can turn into SCD [52]. In the same study, 81.6% were able to identify that SCD is a genetic disease [52]. Focus groups within the same study identified lack of education in the community as a major barrier to more widespread awareness of SCT [52]. In the university-based study, 58% (n = 150) incorrectly believed that trait causes many health problems, 41% (n = 107) believed that SCT causes many deaths, and 37% (n = 96) believed that SCT causes crisis [53].
From the provider perspective, adult providers seem to prefer screening with the SCST compared to pediatricians. Two studies examined providers’ preferences for screening for SCT utilizing web-based surveys following the implementation of the NCAA screening protocols [55, 56]. In the pediatrician survey, 58% of pediatricians preferred the SCST compared with 71% preferring to use the medical record and 60% preferring to use newborn screening [55]. In contrast, among 370 members of the American Medical Society for Sports Medicine (AMSSM), 71% favored using the SCST to screen for SCD, versus 72% relying on the medical record and 53% using newborn screening [56]. This may be due to lack of awareness of the test as more pediatricians surveyed were not aware of the SCST (43%) compared to the sports medicine physicians (12%) [55, 56].
| Future Directions Beyond the Solubility Test | ▴Top |
The SCST remains a useful and inexpensive modality for screening for SCD or SCT even after considering its limitations. However, novel technologies for point-of-care testing are on the horizon and these may overcome the limitations of the SCST by offering more accurate and rapid diagnosis. Various new technologies have been developed including paper-based hemoglobin solubility, lateral flow immunoassays, micro-engineered HE, density-based separation, and smartphone-based application tests [57]. Of these, the lateral flow immunoassays have gained the most traction in resource-poor settings. The two most widely available assays are the Sickle SCAN® (BioMedomics Inc., Morrisville, NC, USA) and the HemoTypeSC® (Silver Lake Research, Azusa, CA, USA) [58]. These assays rely on the formation antibody-antigen complexes between commercially available antibodies and patient supplied antigens (hemoglobin), and thus can distinguish between SCD and SCT [58]. The Sickle SCAN® uses polyclonal antibody targets against HbA, HbS and hemoglobin C (HbC) to identify the hemoglobin type, with a positive test indicated by a line next to the corresponding hemoglobin type [57, 58]. In comparison, the HemoTypeSC® utilizes monoclonal antibodies against HbA, HbS and HbC on fixed test lines, with a positive result indicated by the absence of a line next to the hemoglobin type [57, 58]. Both tests can be performed on infants with high HbF and are inexpensive (less than $5) [58]. In a study that prospectively analyzed the accuracy of the Sickle SCAN® and the HemoTypeSC® in 1,000 blood samples each in Angola, both correlated 100% to isoelectric focusing, with the HemoTypeSC® requiring longer time to run (15 min vs. 7 min) and more repeats due to invalid results (68 times vs. 2 times for Sickle SCAN®) [59]. These technologies are likely to eventually supplant the SCST.
Acknowledgments
We would like to acknowledge Inova Blood Donor Services for their help in preparing the samples for Figure 1.
Financial Disclosure
None to declare.
Conflict of Interest
Sebastian Mendez-Marti, MD has no conflict of interest to disclose. Chad Zik, MD has no conflict of interest to disclose. Shenei Alan, MD PhD receives research funding through Pfizer and participates in the Pfizer speaker’s bureau. Hongkun Wang, PhD has no conflict of interest to disclose. William B. Ershler, MD serves on the advisory board for Pfizer, Novartis and Pharmacosmos. He receives research funding and participates in the Pfizer speaker’s bureau.
Author Contributions
SMM performed the data collection, prepared the figures, prepared, and edited the manuscript. CZ also performed data collection and edited the manuscript. SA co-wrote and edited the manuscript. HW aided in the study design, statistical analysis, and edited the manuscript. WBE designed the data collection, co-wrote and edited the manuscript. All authors read and approved the final manuscript.
Data Availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
Abbreviations
AMSSM: American Medical Society for Sports Medicine; CDC: Centers for Disease Control and Prevention; HR: hazard ratio; HbF: fetal hemoglobin; HbC: hemoglobin C; HbS: hemoglobin S; HE: hemoglobin electrophoresis; NCAA: National Collegiate Athletic Association; NHLBI: National Heart, Lung and Blood Institute; NIH: National Institutes of Health; PCPs: primary care providers; SCD: sickle cell disease; SCST: sickle cell solubility test; SCT: sickle cell trait; US: United States; UTPs: universal training precautions
| References | ▴Top |
- Itano HA, Pauling L. A rapid diagnostic test for sickle cell anemia. Blood. 1949;4(1):66-68.
pubmed - Benson JM, Therrell BL, Jr. History and current status of newborn screening for hemoglobinopathies. Semin Perinatol. 2010;34(2):134-144.
doi pubmed - Canning DM, Huntsman RG. An assessment of Sickledex as an alternative to the sickling test. J Clin Pathol. 1970;23(8):736-737.
doi pubmed pmc - Nalbandian RM, Nichols BM, Camp FR, Jr., Lusher JM, Conte NF, Henry RL, Wolf PL. Dithionite tube test—a rapid, inexpensive technique for the detection of hemoglobin S and non-S sickling hemoglobin. Clin Chem. 1971;17(10):1028-1032.
pubmed - Tubman VN, Field JJ. Sickle solubility test to screen for sickle cell trait: what's the harm? Hematology Am Soc Hematol Educ Program. 2015;2015:433-435.
doi pubmed - Hicksg EJ, Griep JA, Nordschow CD. Comparison of results for three method of hemoglobin S identification. Clin Chem. 1973;19(5):533-535.
pubmed - 005223: Hemoglobin (Hb) Solubility | Labcorp. Accessed May 22, 2024. https://www.labcorp.com/tests/005223/hemoglobin-hb-solubility.
- PubChem. Sodium Dithionite. Accessed May 22, 2024. https://pubchem.ncbi.nlm.nih.gov/compound/24489.
- Okwi AL, Ocaido M, Byarugaba W, Ndugwa CM, Parkes A. Sickling and solubility tests and the peripheral blood film method for screening for sickle cell disease. [corrected]. S Afr Med J. 2009;99(12):887-891.
pubmed - Arishi WA, Alhadrami HA, Zourob M. Techniques for the Detection of Sickle Cell Disease: A Review. Micromachines (Basel). 2021;12(5).
doi pubmed pmc - Kato GJ, Piel FB, Reid CD, Gaston MH, Ohene-Frempong K, Krishnamurti L, Smith WR, et al. Sickle cell disease. Nat Rev Dis Primers. 2018;4:18010.
doi pubmed - Gaston MH, Verter JI, Woods G, Pegelow C, Kelleher J, Presbury G, Zarkowsky H, et al. Prophylaxis with oral penicillin in children with sickle cell anemia. A randomized trial. N Engl J Med. 1986;314(25):1593-1599.
doi pubmed - Consensus conference. Newborn screening for sickle cell disease and other hemoglobinopathies. JAMA. 1987;258(9):1205-1209.
pubmed - Pinto VM, De Franceschi L, Gianesin B, Gigante A, Graziadei G, Lombardini L, Palazzi G, et al. Management of the Sickle Cell Trait: An Opinion by Expert Panel Members. J Clin Med. 2023;12(10):3441.
doi pubmed pmc - Ojodu J, Hulihan MM, Pope SN, Grant AM, Centers for Disease C, Prevention. Incidence of sickle cell trait—United States, 2010. MMWR Morb Mortal Wkly Rep. 2014;63(49):1155-1158.
pubmed pmc - Pecker LH, Naik RP. The current state of sickle cell trait: implications for reproductive and genetic counseling. Blood. 2018;132(22):2331-2338.
doi pubmed pmc - Naik RP, Haywood C, Jr. Sickle cell trait diagnosis: clinical and social implications. Hematology Am Soc Hematol Educ Program. 2015;2015(1):160-167.
doi pubmed pmc - Taylor SM, Parobek CM, Fairhurst RM. Haemoglobinopathies and the clinical epidemiology of malaria: a systematic review and meta-analysis. Lancet Infect Dis. 2012;12(6):457-468.
doi pubmed pmc - Tsaras G, Owusu-Ansah A, Boateng FO, Amoateng-Adjepong Y. Complications associated with sickle cell trait: a brief narrative review. Am J Med. 2009;122(6):507-512.
doi pubmed - Ashcroft MT, Desai P. Mortality and morbidity in Jamaican adults with sickle-cell trait and with normal haemoglobin followed up for twelve years. Lancet. 1976;2(7989):784-786.
doi pubmed - Ataga KI, Saraf SL, Derebail VK. The nephropathy of sickle cell trait and sickle cell disease. Nat Rev Nephrol. 2022;18(6):361-377.
doi pubmed pmc - Gupta AK, Kirchner KA, Nicholson R, Adams JG, 3rd, Schechter AN, Noguchi CT, Steinberg MH. Effects of alpha-thalassemia and sickle polymerization tendency on the urine-concentrating defect of individuals with sickle cell trait. J Clin Invest. 1991;88(6):1963-1968.
doi pubmed pmc - Iacovelli R, Modica D, Palazzo A, Trenta P, Piesco G, Cortesi E. Clinical outcome and prognostic factors in renal medullary carcinoma: A pooled analysis from 18 years of medical literature. Can Urol Assoc J. 2015;9(3-4):E172-177.
doi pubmed pmc - Jones SR, Binder RA, Donowho EM, Jr. Sudden death in sickle-cell trait. N Engl J Med. 1970;282(6):323-325.
doi pubmed - Webber BJ, Witkop CT. Screening for sickle-cell trait at accession to the United States military. Mil Med. 2014;179(11):1184-1189.
doi pubmed - Nye NS, Grubic T, Kim M, O'Connor F, Deuster PA. Universal Training Precautions: A Review of Evidence and Recommendations for Prevention of Exercise-Related Injury, Illness, and Death in Warfighters and Athletes. J Athl Train. 2023;58(3):232-243.
doi pubmed pmc - Connes P, Hyacinth HI, Naik RP. Sickle cell trait. In: Gladwin MT, Kato GJ, Novelli EM, eds. Sickle cell disease. McGraw-Hill Education; 2021. Accessed May 22, 2024. hemonc.mhmedical.com/content.aspx?aid=1179344404.
- Mooney B. Army begins screening recruits for sickle cell trait, joining other services. U.S. Medicine. Published May 11, 2021. Accessed May 22, 2024. https://www.usmedicine.com/2021-compendium-of-federal-medicine/army-begins-screening-recruits-for-sickle-cell-trait-joining-other-services/.
- Kavanagh PL, Fasipe TA, Wun T. Sickle Cell Disease: A Review. JAMA. 2022;328(1):57-68.
doi pubmed - Tarini BA, Brooks MA, Bundy DG. A policy impact analysis of the mandatory NCAA sickle cell trait screening program. Health Serv Res. 2012;47(1 Pt 2):446-461.
doi pubmed pmc - Parsons JT. 2014-2015 NCAA Sports Medicine Handbook. National Collegiate Athletic Association; 2014. https://www.ncaapublications.com/productdownloads/MD15.pdf.
- 2023-2024 NCAA Division I Manual. National Collegiate Athletic Association; 2023. Accessed October 3, 2023. https://www.ncaapublications.com/p-4673-2023-2024-ncaa-division-i-manual.aspx.
- 2023-2024 NCAA Division II Manual. National Collegiate Athletic Association; 2023. Accessed October 3, 2023. https://www.ncaapublications.com/p-4674-2023-2024-ncaa-division-ii-manual.aspx.
- 2023-2024 NCAA Division III Manual. National Collegiate Athletic Association; 2023. Accessed October 3, 2023. https://www.ncaapublications.com/p-4675-2023-2024-ncaa-division-iii-manual.aspx.
- McDonald MA, Creary MS, Powell J, Daley LA, Baker C, Royal CD. Perspectives and Practices of Athletic Trainers and Team Physicians Implementing the 2010 NCAA Sickle Cell Trait Screening Policy. J Genet Couns. 2017;26(6):1292-1300.
doi pubmed - Sickle cell trait. NCAA.org. Accessed May 22, 2024. https://www.ncaa.org/sports/2016/7/27/sickle-cell-trait.aspx.
- Bain BJ, Daniel Y, Henthorn J, de la Salle B, Hogan A, Roy NBA, Mooney C, et al. Significant haemoglobinopathies: A guideline for screening and diagnosis: A British Society for Haematology Guideline: A British Society for Haematology Guideline. Br J Haematol. 2023;201(6):1047-1065.
doi pubmed - Frequently asked questions. Accessed June 5, 2024. https://www.redcrossblood.org/faq.html.
- Schuetz AN, Hillyer KL, Roback JD, Hillyer CD. Leukoreduction filtration of blood with sickle cell trait. Transfus Med Rev. 2004;18(3):168-176.
doi pubmed - Gehrie EA, Petran L, Young PP. Sickle cell trait results in a high leukoreduction quality control failure rate for whole blood donations. Transfusion. 2022;62(9):1727-1730.
doi pubmed pmc - Singer DE, Byrne C, Chen L, Shao S, Goldsmith J, Niebuhr DW. Risk of Exertional Heat Illnesses Associated with Sickle Cell Trait in U.S. Military. Mil Med. 2018;183(7-8):e310-e317.
doi pubmed pmc - Nelson DA, Deuster PA, Carter R, 3rd, Hill OT, Wolcott VL, Kurina LM. Sickle Cell Trait, Rhabdomyolysis, and Mortality among U.S. Army Soldiers. N Engl J Med. 2016;375(5):435-442.
doi pubmed pmc - Nelson DA, Deuster PA, O'Connor FG, Kurina LM. Sickle Cell Trait and Heat Injury Among US Army Soldiers. Am J Epidemiol. 2018;187(3):523-528.
doi pubmed - DeBaun M. Universal precautions help decrease rate of exercise-related death in patient with sickle cell trait. The Hematologist. 2016;13(6).
doi - Nelson SC. Scaring athletes with sickle cell trait. Am J Cardiol. 2013;111(1):149.
doi pubmed - Harris KM, Haas TS, Eichner ER, Maron BJ. Sickle cell trait associated with sudden death in competitive athletes. Am J Cardiol. 2012;110(8):1185-1188.
doi pubmed - Maron BJ, Doerer JJ, Haas TS, Tierney DM, Mueller FO. Sudden deaths in young competitive athletes: analysis of 1866 deaths in the United States, 1980-2006. Circulation. 2009;119(8):1085-1092.
doi pubmed - Eichner ER. Sickle cell trait in sports. Curr Sports Med Rep. 2010;9(6):347-351.
doi pubmed - Statement on screening for sickle cell trait and athletic participation. Accessed May 22, 2024. https://www.hematology.org/advocacy/policy-news-statements-testimony-and-correspondence/policy-statements/2012/screening-sickle-cell-trait-athletic-participation.
- Thompson AA. Sickle cell trait testing and athletic participation: a solution in search of a problem? Hematology Am Soc Hematol Educ Program. 2013;2013:632-637.
doi pubmed - Kavanagh PL, Wang CJ, Therrell BL, Sprinz PG, Bauchner H. Communication of positive newborn screening results for sickle cell disease and sickle cell trait: variation across states. Am J Med Genet C Semin Med Genet. 2008;148C(1):15-22.
doi pubmed - Treadwell MJ, McClough L, Vichinsky E. Using qualitative and quantitative strategies to evaluate knowledge and perceptions about sickle cell disease and sickle cell trait. J Natl Med Assoc. 2006;98(5):704-710.
pubmed pmc - Harrison SE, Walcott CM, Warner TD. Knowledge and Awareness of Sickle Cell Trait Among Young African American Adults. West J Nurs Res. 2017;39(9):1222-1239.
doi pubmed - Adigwe OP. Knowledge and awareness of sickle cell disease: a cross sectional study amongst unmarried adults in Nigeria's capital city. J Community Genet. 2022;13(6):579-585.
doi pubmed pmc - Koopmans J, Cox LA, Benjamin H, Clayton EW, Ross LF. Sickle cell trait screening in athletes: pediatricians' attitudes and concerns. Pediatrics. 2011;128(3):477-483.
doi pubmed - Acharya K, Benjamin HJ, Clayton EW, Ross LF. Attitudes and beliefs of sports medicine providers to sickle cell trait screening of student athletes. Clin J Sport Med. 2011;21(6):480-485.
doi pubmed - Kawooya I, Kayongo E, Munube D, et al. Point-of-care diagnostic tests for sickle cell disease. Cochrane Database Syst Rev. 2022;2022(9).
doi - Dexter D, McGann PT. Saving lives through early diagnosis: the promise and role of point of care testing for sickle cell disease. Br J Haematol. 2022;196(1):63-69.
doi pubmed - Olaniyan HS, Briscoe C, Santos B, Pascoal R, Armando A, McGann PT. Comparison of sickle SCAN and hemotype SC as point-of-care newborn screening diagnostics for sickle cell disease in Luanda, Angola. Blood. 2021;138(Supplement 1):913.
doi
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.