

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://www.thejh.org |
Case Report
Volume 13, Number 5, October 2024, pages 229-237

Systemic Epstein-Barr Virus-Positive T-Cell Lymphoma of Childhood Associated With t(1;22)(p22;q11.2) Mutation
Lane Lernera, e, Sushanth Sreenivasanb, e, f, Chelsea Petersonc, Maitreyee Raic, Pragnan Kancharlac, Samuel Santosad, Mark Bunkerd, Yazan Samhouric
aCollege of Medicine, Unit K, Drexel University, Philadelphia, PA, USA
bDepartment of Medicine, Allegheny General Hospital, Pittsburgh, PA, USA
cDivision of Hematology and Cellular Therapy, Allegheny Health Network Cancer Institute, Pittsburgh, PA, USA
dDivision of Pathology, Allegheny General Hospital, Pittsburgh, PA, USA
eThese authors contributed equally to this article.
fCorresponding Author: Sushanth Sreenivasan, Department of Medicine, Allegheny General Hospital, Pittsburgh, PA 15224, USA
Manuscript submitted May 15, 2024, accepted September 17, 2024, published online October 3, 2024
Short title: Systemic EBV+ T-Cell Lymphoma of Childhood
doi: https://doi.org/10.14740/jh1284
| Abstract | ▴Top |
Systemic Epstein-Barr virus-positive (EBV+) T-cell lymphoma (TCL) of childhood is an uncommon TCL that occurs secondary to an acute or chronic EBV infection. The disorder is characterized by the monoclonal expansion of EBV+ T cells driven by an increased immune response and defect in regulatory pathways. Thus, systemic EBV+ TCL of childhood is frequently associated with a hyperinflammatory state, hemophagocytic lymphohistiocytosis (HLH) syndrome, and exhibits a fulminant clinical course with poor outcomes. Additionally, genetic alterations at specific chromosome loci, such as chromosome 22q11.2, are hypothesized to increase the chances of carcinogenic transformation and increase the risk of non-Hodgkin lymphoma later in life. Chemotherapy, immunotherapy, and allogenic stem cell transplants are treatment options with varying degrees of success. In this report, we describe a case of a 21-year-old male with a primary acute EBV infection that led to HLH syndrome. He was ultimately diagnosed with systemic EBV+ TCL of childhood. Despite treatment chemotherapy, the patient passed before an allogenic stem cell transplant could be performed. We explore the clinicopathological features of his disease and a possible new oncogenic locus at the t(1;22)(p22;q11.2) breakpoint. Our case underscores the importance of retaining a wide differential diagnosis, including unusual presentations of systemic EBV+ TCL of childhood, when presented with an adult case of HLH. It also highlights a possible new genetic locus associated with immunological malignancies that warrants further study.
Keywords: Systemic EBV-positive T-cell lymphoma of childhood; t(1;22)(p22;q11.2); Epstein-Barr virus; Hemophagocytic lymphohistiocytosis syndrome
| Introduction | ▴Top |
Systemic Epstein-Barr virus-positive (EBV+) T-cell lymphoma (TCL) of childhood is a rare disease with varying presentations, including hemophagocytic lymphohistiocytosis (HLH) syndrome, severe sepsis, or multiorgan failure in a clinical setting. The disorder is characterized by the monoclonal expansion of EBV+ T cells with an activated cytotoxic phenotype [1, 2]. Most cases of both systemic chronic active EBV disease (CAEBV) and systemic EBV+ TCL of childhood occur in pediatric and adolescent Asian populations - particularly in Japan, Taiwan, and China - and Native American, Central and South American populations [3]. Here, we describe a rare, fatal case of EBV+ TCL with secondary HLH in a White male patient.
The clonal expansion of EBV+ T cells with cytotoxic phenotype is driven by an increased immune response to persistent EBV+ infected cells, which results in excessive proliferation of T cells [2]. The process is further aided by disruption of regulatory immune pathways that would temper the body’s immune response [4]. Thus, systemic EBV+ TCL of childhood is frequently associated with a hyperinflammatory state and exhibits a fulminant clinical course.
Histopathological characteristics of the systemic EBV+ TCL include bone marrow, spleen and liver infiltration with small lymphoid cells with histiocytic hyperplasia, and hemophagocytosis [5]. These neoplastic lymphoid cells are indistinguishable from normal lymphocytes and can show minimal to no cellular atypia; they are EBV+ CD8 cytotoxic T cells. These cells also express CD2, CD3, TIA-1, and granzyme B, but are negative for CD56 expression. Rare cases exhibit both CD4- and CD8-positive T cells. LMP1 and EBNA2, viral proteins essential for cellular transformation, are commonly absent in neoplastic cells following EBV infection. Infiltrating T cells generally have monoclonal rearrangements of the T-cell receptor genes, and EBV is present in a clonal episomal form [3]. The serological tests to diagnose active EBV infection are inconsistent and nondiagnostic, anti-viral capsid antigen IgM (VCA) is often negative, while IgG VCA antibodies are generally positive. On the other hand, the EBV in situ hybridization (ISH) testing can confirm the EBV infection [3].
There are several genetic alterations that can predispose these patients to carcinogenic transformation following frequent and intensive infection from the EBV. Genetic deletions at chromosome locus 22q11.2 have been associated with DiGeorge syndrome, velocardiofacial syndrome, and the Cayler cardiofacial syndrome [6]. In all these instances, the disorders are characterized by thymic hypoplasia, leading to a range of T-cell deficits and interference with tumor surveillance [6]. Consequently, these patients have an increased predisposition for non-Hodgkin lymphoma [7].
In this case report, we describe the clinicopathological features of systemic EBV+ TCL of childhood that arose in the setting of HLH secondary to EBV infection, review treatment options, and investigate the meaning of a possible constitutional t(1;22)(p22;q11.2) mutation.
| Case Report | ▴Top |
A 21-year-old Caucasian male was hospitalized in February of 2022 for intermittent fevers, chest pain, shortness of breath, fatigue, and tachycardia of 1 week duration. His past medical history was negative for severe mosquito bite allergy, DiGeorge syndrome or other disorders associated with chromosome locus 22q11.2. Upon physical examination, he presented with hepatosplenomegaly and tachycardia. Laboratory findings established a decreased white blood cell (WBC) count of 1.44 × 109/L, a lactic acid level of 3.0 mmol/L, and a ferritin level of 16,782 ng/mL. An electrocardiogram (EKG) showed sinus tachycardia. A computed tomography (CT) scan of the thorax, abdomen, and pelvis exhibited lymphadenopathy in the periaortic and lower mesenteric areas. A biopsy of the left inguinal lymph node found atypical cells with prominent erythrophagocytosis. A bone marrow biopsy showed mild, interstitial T-cell infiltrate with focal EBV+ cells, which were identified by ISH. There was no evidence of hemophagocytosis on bone marrow biopsy. A fluorescence in situ hybridization (FISH) study showed the patient possessed a t(1;22)(p22;q11.2) translocation. An infectious mononucleosis screen showed Q-PCR EBV levels of 10,400 International Unit (IU) per mL, soluble interleukin-2 receptor (IL-2R) alpha testing was 24,412 U/mL (reference range: 223 - 710). TCR rearrangement was not detected by PCR testing for beta and gamma rearrangements. HTLV I/II antibody testing was also negative. The patient was diagnosed with HLH, secondary to EBV infection.
The patient was treated with intravenous methylprednisolone, intravenous immune globulin (IVIG), and weekly rituximab for four doses, but his symptoms persisted. EBV titers peaked at 39,400 IU/mL. Eventually, 10 doses of anakinra were added to his treatment regimen, and his symptoms of fever, tachycardia, hypoxia, and pain resolved. He was discharged on prednisone and EBV titers trended down to 360.0 IU/mL. Unfortunately, his EBV titers started to trend up again. The fevers returned 3 months later, and he was admitted for concerns of disease progression. The patient once again presented with marked hepatosplenomegaly and fever. The patient reported nausea, fatigue, and a sore throat. Laboratory findings showed marked pancytopenia (WBC 1.55 × 109/L, hemoglobin 12.8 × 109/L, absolute neutrophil count 1.01 × 109/L, and absolute lymphocyte count 0.36 × 109/L), EBV Q-PCR of 3,850.0 copies, high ferritin of 19,687 ng/mL, triglyceride level of 167 mmol/L, IL-8 level of 8.7 pg/mL (reference range 0.0 - 34.7) and an IL-2R alpha level of 2,211 U/mL. He fulfilled 6/8 criteria for the diagnosis of HLH: fever, splenomegaly, hemophagocytosis in the lymph nodes, absent natural killer (NK) cell activity, elevated IL-2 levels > 2,400 U/mL and serum ferritin > 500 ng/mL.
A CT of the thorax, abdomen, and pelvis revealed innumerable pulmonary nodules and hypodense lesions in the liver and spleen. Magnetic resonance imaging (MRI) of liver further defined these lesions as large rim enhancing lesions permeating throughout the liver and spleen, and some lesions had delayed continuous peripheral contrast fill-in. Due to suspicion of lymphoma, progressive HLH, or a diffuse fungal infection, biopsies were taken from a pulmonary nodule, the left inguinal lymph node, a liver lesion, and bone marrow.
After coordinating with the National Institutes of Health (NIH) to analyze the pathological slides, the bone marrow biopsy showed mildly hypocellular bone marrow, with interstitial T-lymphocytic infiltrate, negative for EBV+ cells. Next-generation sequencing performed for a broad array of mutations detected in lymphoid disorders was negative.
The left inguinal lymph node showed effacement of its architecture by atypical lympho-histiocytic infiltrates, predominantly by CD3+ T cells and few scattered CD20+ B cells (Figs. 1-3). These infiltrates were composed of atypical lymphoid cells mixed with numerous histiocytes displaying prominent erythrophagocytosis (Figs. 4 and 5).
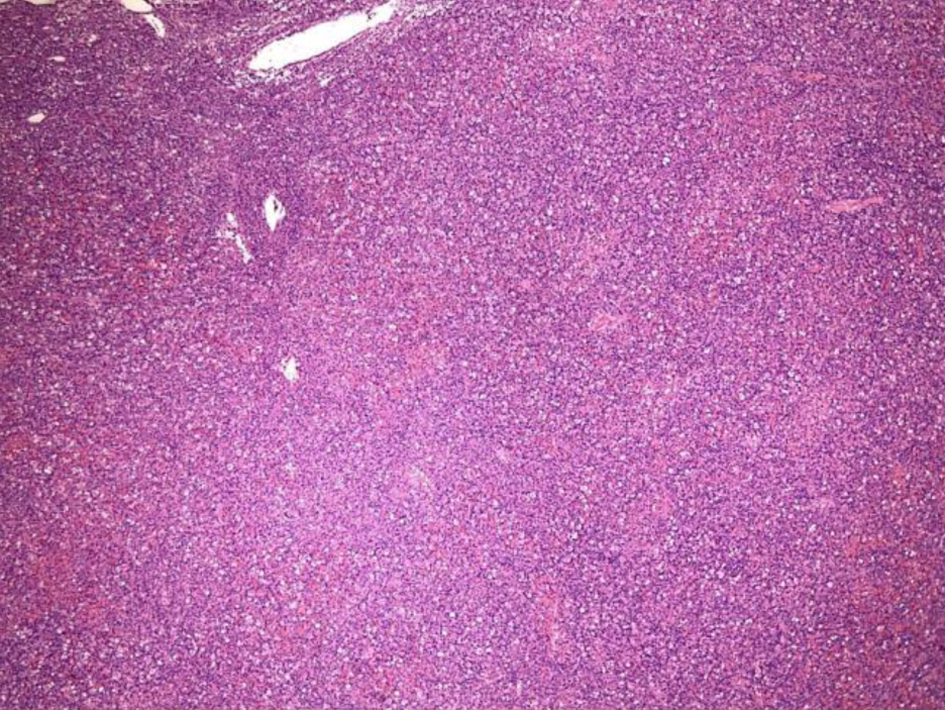 Click for large image | Figure 1. Low-power view (× 4) of left inguinal lymph node from an excisional biopsy that shows effacement of the lymph node architecture. |
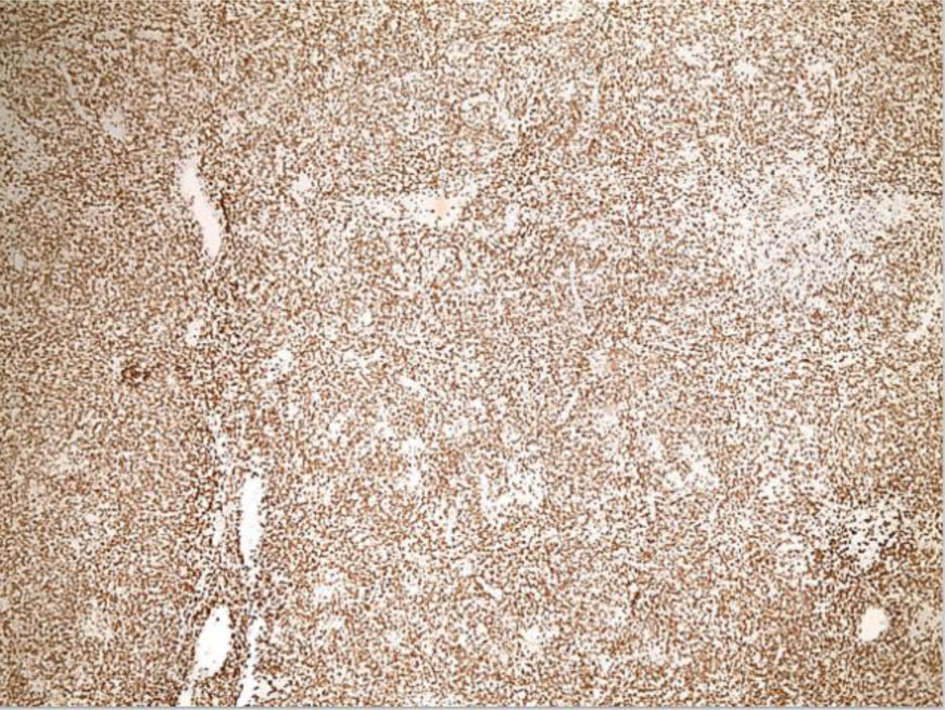 Click for large image | Figure 2. Low-power view (× 4) of left inguinal lymph node from an excisional biopsy after CD3 staining shows effacement of the lymph node architecture is predominantly by CD3+ T cells. |
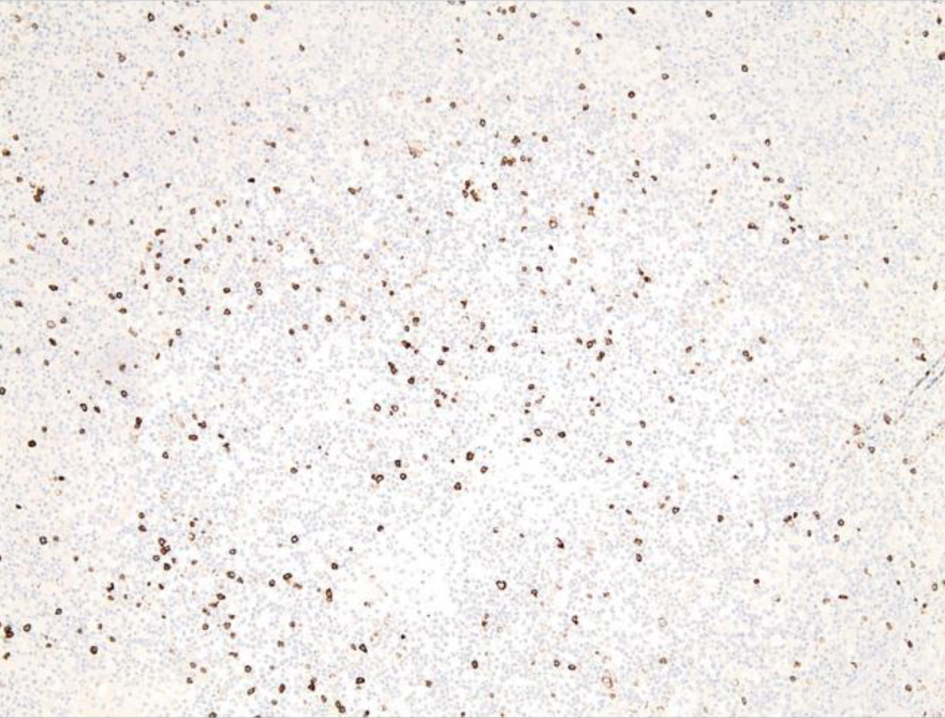 Click for large image | Figure 3. Low-power view of left inguinal lymph node from an excisional biopsy after CD20 staining shows scattered CD20+ B cells. |
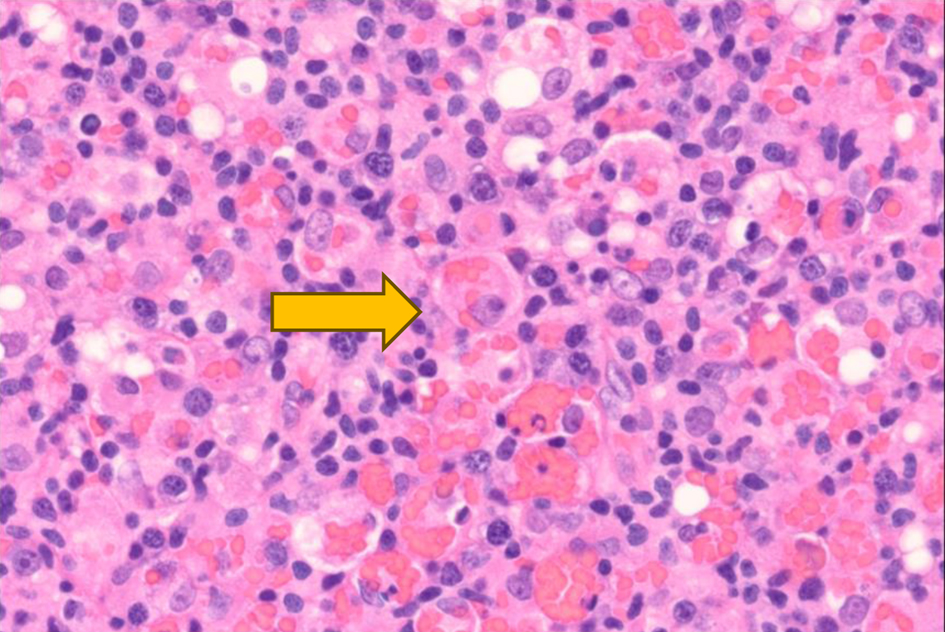 Click for large image | Figure 4. High-power view (× 60) of left inguinal lymph node after hematoxylin and eosin staining reveals histocytes with engulfed red blood cells (× 60) as indicated by the yellow arrow. |
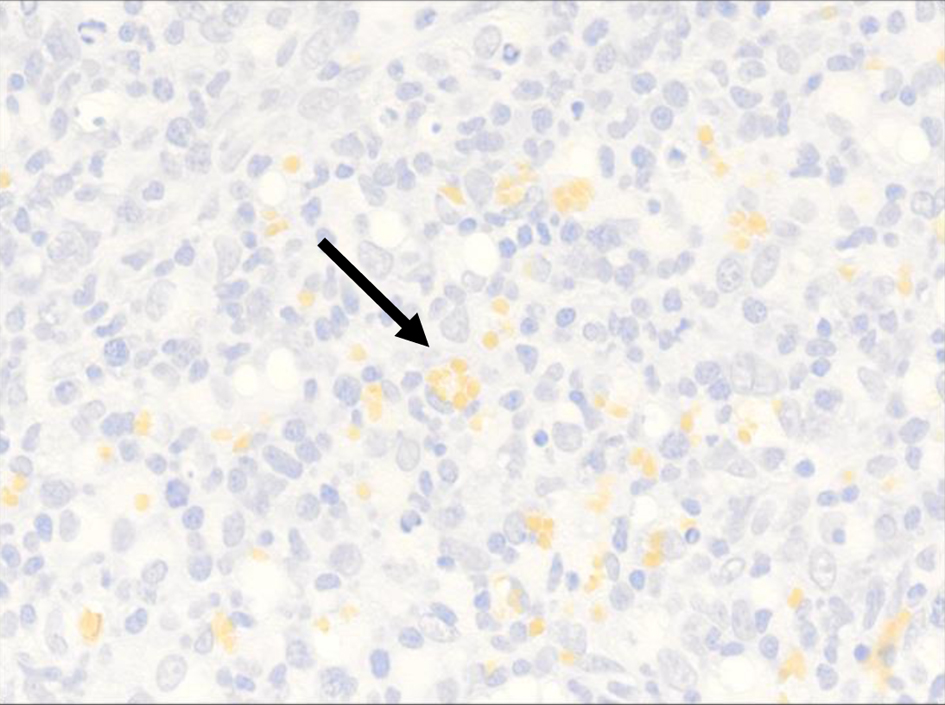 Click for large image | Figure 5. High-power view (× 40) of CD235 highlights intracytoplasmic red blood cells within the histiocyte (black arrow). |
The lung nodule biopsy was largely necrotic with few viable areas. The viable regions contained an atypical lympho-histiocytic infiltrate, predominantly composed of CD3+ T cells (Figs. 6 and 7). Epstein-Barr encoding region ISH, a method used to detect EBV in tissue sections, showed few scattered positive cells ranging in size (Fig. 8).
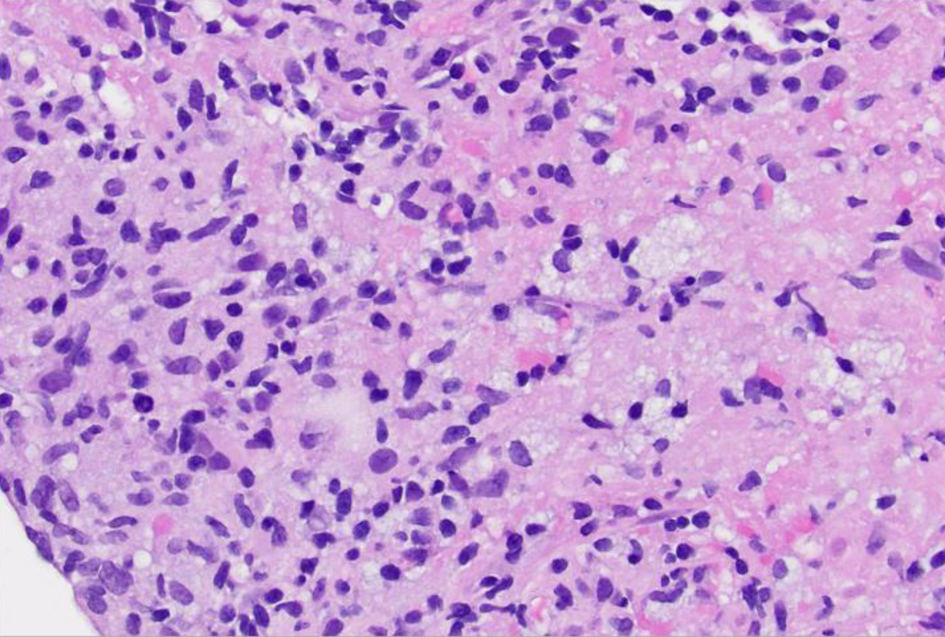 Click for large image | Figure 6. High-power view (× 40) of left lung lesion transbronchial biopsy after hematoxylin and eosin staining reveals infiltrate composed of atypical lymphocytes, some with prominent nucleoli. |
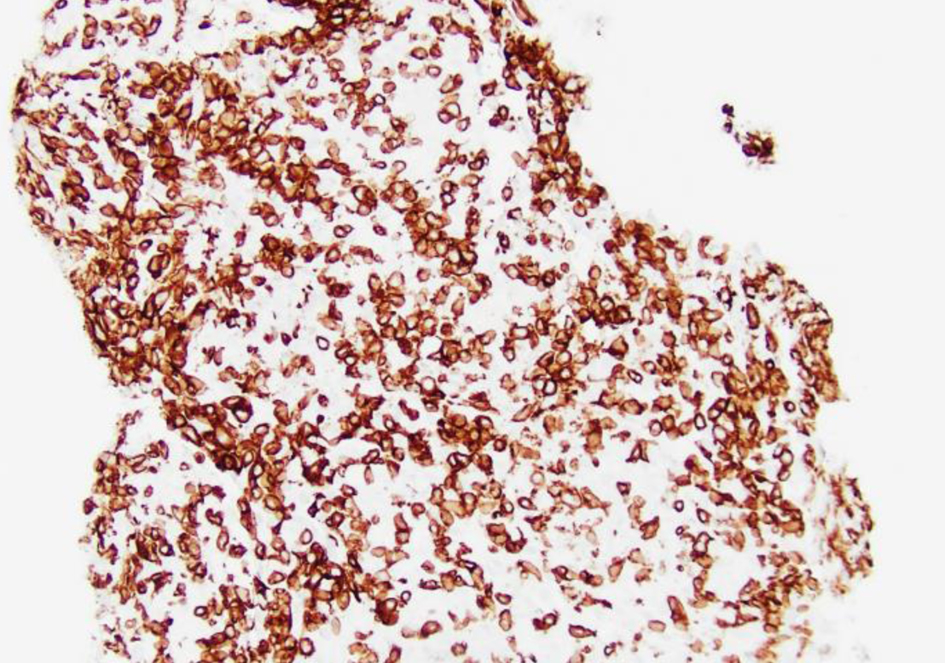 Click for large image | Figure 7. Low-power view (× 20) of left lung lesion biopsy after CD3 staining shows atypical lymphocytic infiltrate predominantly composed of CD3+ T cells. |
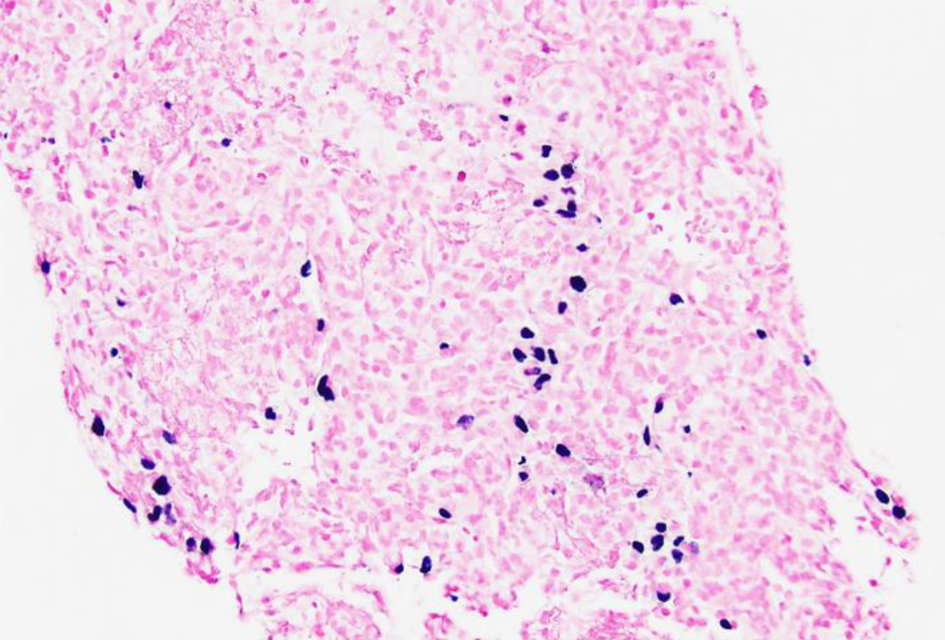 Click for large image | Figure 8. Low-power view (× 20) of left lung lesion biopsy of Epstein-Barr virus (EBV) detection by in situ hybridization (EBER) stain showing that focal large cells within the atypical lymphocytic infiltrate are positive for EBV. |
The liver biopsy also showed atypical dense lympho-histiocytic infiltrates composed of histiocytes and atypical medium to large lymphoid cells (Fig. 9). Approximately 40% of the sample was necrotic. An immunohistochemistry (IHC) staining of the liver biopsy showed atypical lymphocytes identified as a mixture of CD4 and CD8 T cells, with CD4 T cell predominance. They further stained positive for CD2, CD3, CD5, GATA3 (Fig. 10) and negative for anaplastic lymphoma kinase (ALK), CD56 (NK cells) (Fig. 11), and CD1a (dendritic cells). Further CD68 and CD163 staining revealed numerous histiocytes and Kupffer cells in the liver biopsy. An analysis of CD30 (member of the tumor necrosis factor receptor superfamily) and MUM1 (protein expressed in activated T cells) IHC staining showed scattered positive cells in a similar distribution. CD15 (myeloid cells) and CD20 (B-cell marker) stains were negative (Fig. 12), and PAX5 (B-cell transcription factor) stains revealed rare B cells. EBV ISH did indicate scattered EBV+ cells within the liver parenchyma (Fig. 13).
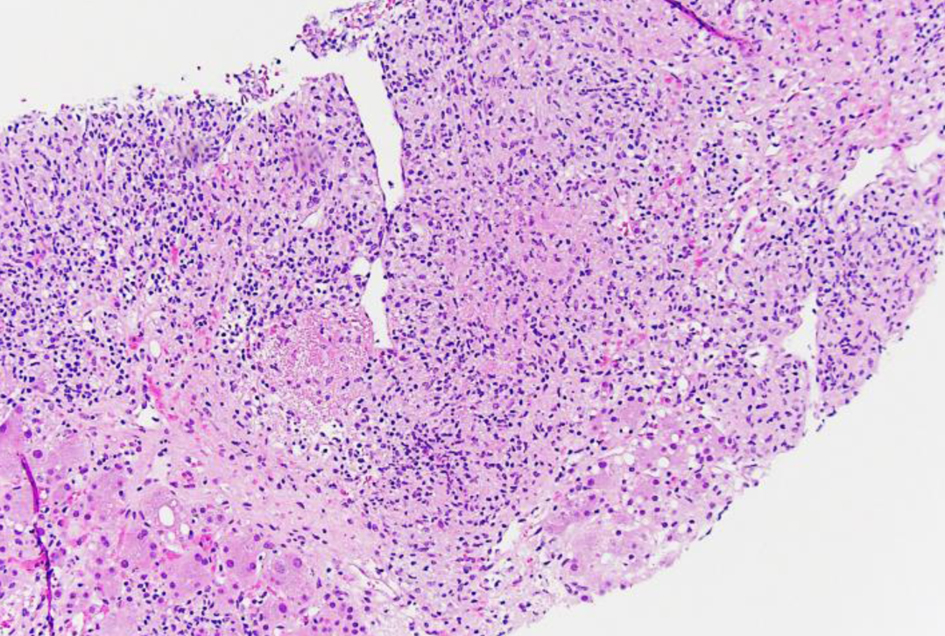 Click for large image | Figure 9. Low-power view (× 10) of liver core biopsy by hematoxylin and eosin stain showing infiltrate composed of atypical lymphocytes with adjacent viable liver. |
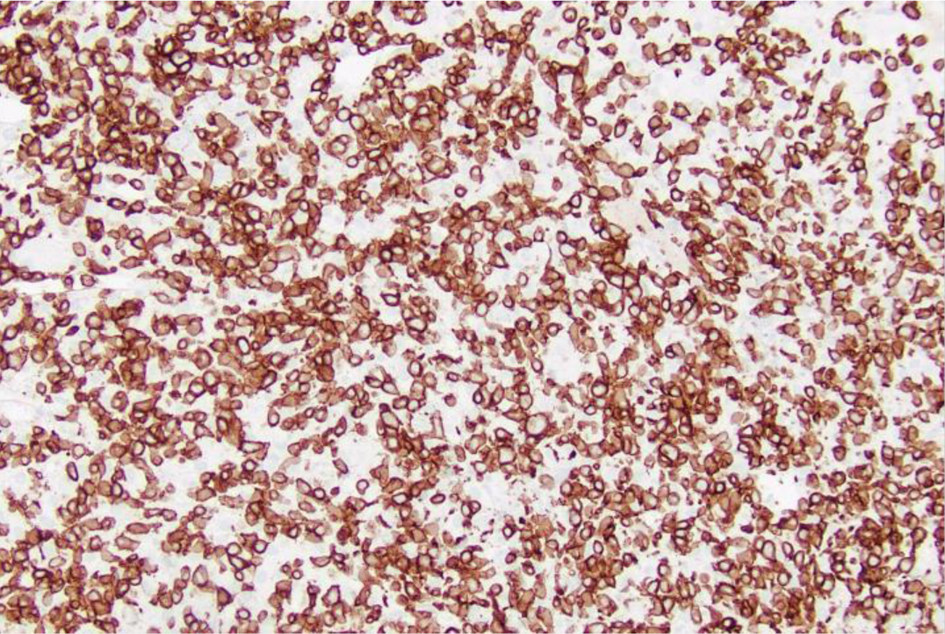 Click for large image | Figure 10. Low-power view (× 20) of liver core biopsy showing CD3+ staining is strongly positive indicating presence of numerous T cells within atypical lymphoid infiltrate. |
 Click for large image | Figure 11. Low-power view (× 10) of liver core biopsy showing CD56 marker is negative indicating the lack natural killer (NK) cells. |
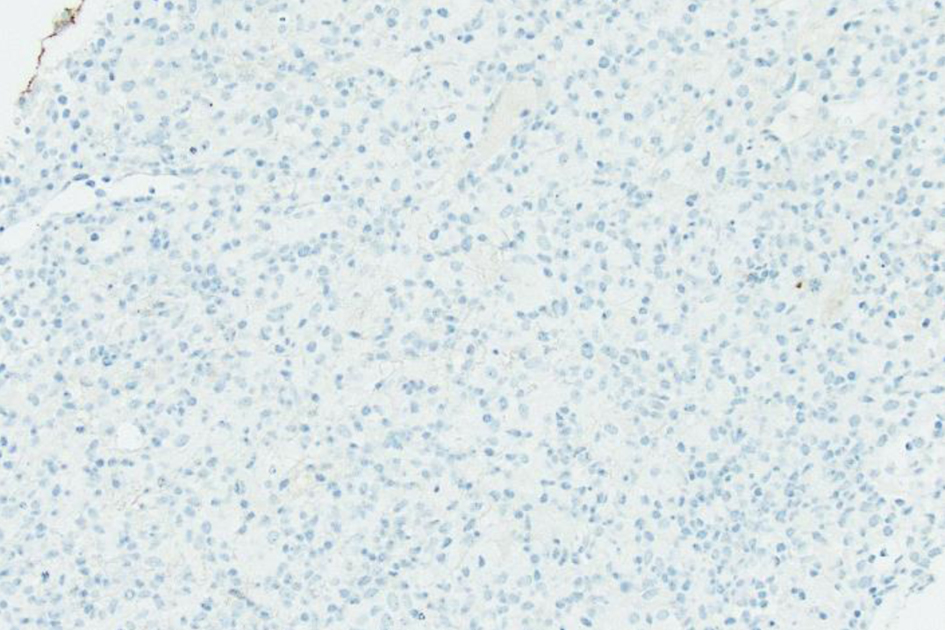 Click for large image | Figure 12. Low-power view (× 20) of liver core biopsy by CD20 stain, which is largely negative, reveals lymphoid infiltrate containing rare B cells. |
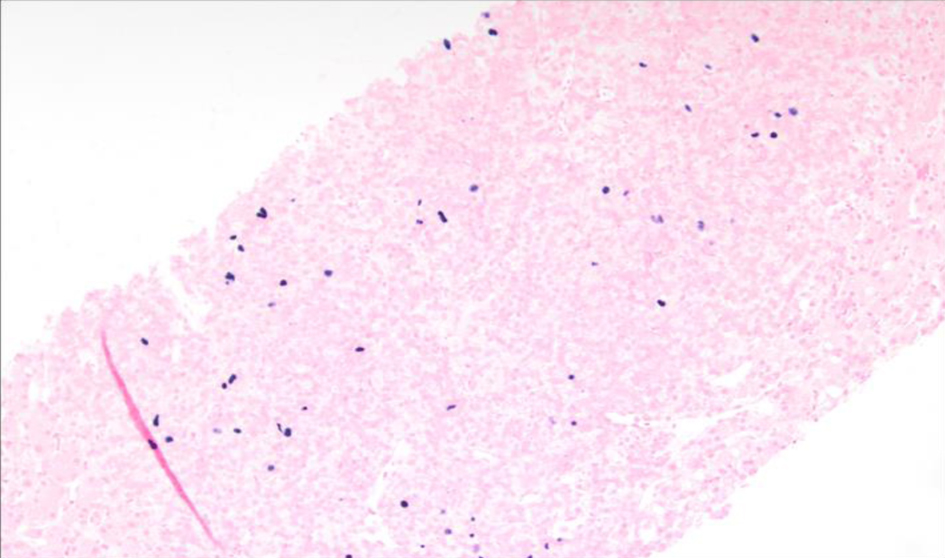 Click for large image | Figure 13. Low-power view (× 10) of Epstein-Barr virus (EBV) in situ hybridization (ISH) or EBV encoded small RNA (EBER) ISH indicating scattered EBV+ cells within the liver parenchyma. |
Cytogenetic FISH studies from the patient’s bone marrow showed an abnormal male karyotype positive for a t(1;22)(p22;q11.2) translocation in every cell of 20 cells found in metaphase, indicating that it could be a constitutional mutation (Fig. 14). He was negative for the most common abnormalities related to non-Hodgkin’s lymphoma and chromosome 21 deletion.
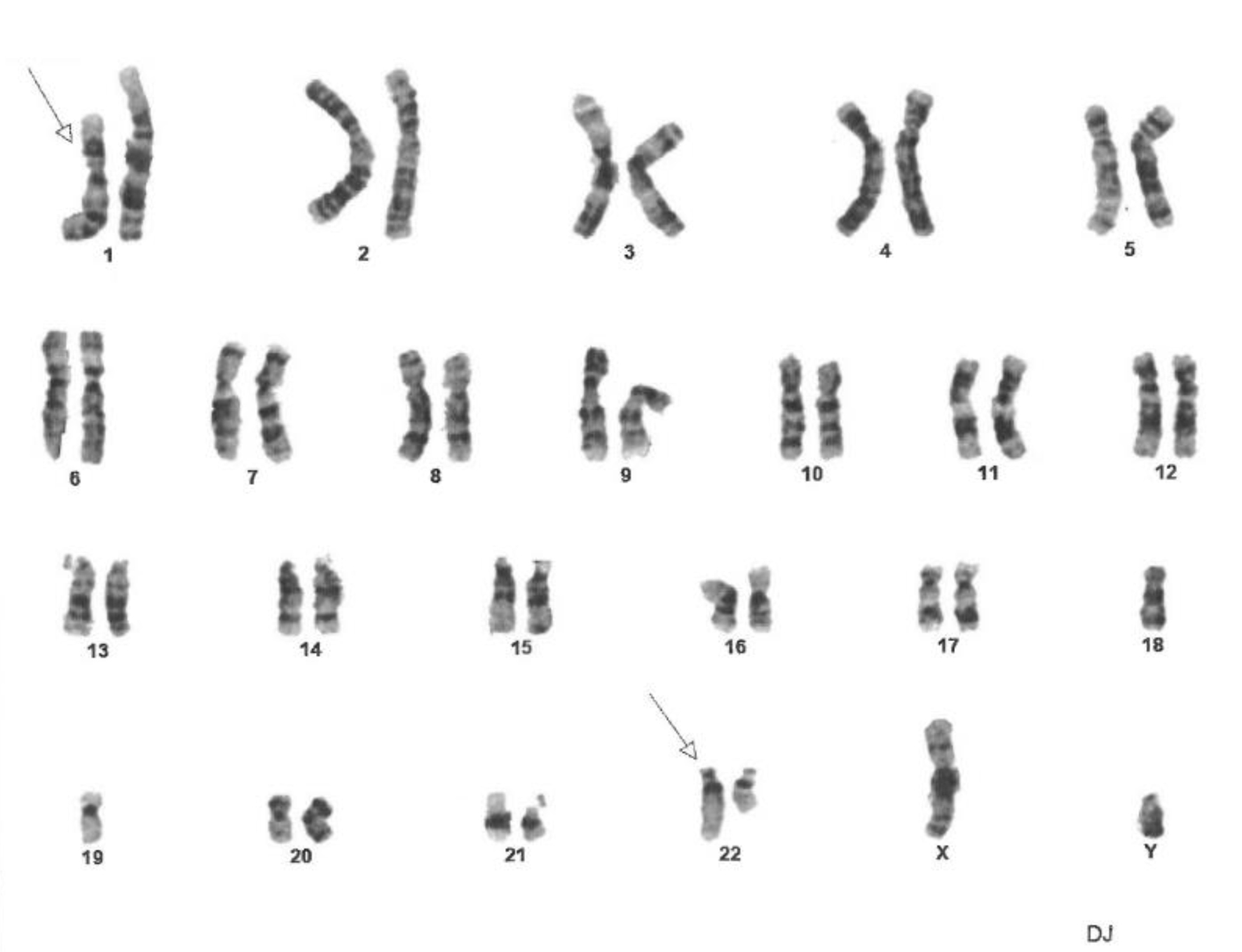 Click for large image | Figure 14. Fluorescence in situ hybridization (FISH) analysis of cells in metaphase showing constitutional mutation t(1;22)(p22;q11.2). The constitutional mutation was found in 100% of cells. Twenty cells in metaphase were analyzed using ASR DNA probes (Abbott Molecular Inc.) for common genetic abnormalities found in acute lymphoblastic leukemia, non-Hodgkin lymphoma and Hodgkin lymphoma. It did not detect any abnormalities on D6Nz1, MYB on chromosome 6, MYC on 8q. D13S319 and LAMP1 on 13q. IGH on 14q. and TP53 on 17p. |
Based on all these tests, it was determined that the pathology was most consistent with systemic EBV+ TCL of childhood. The patient was prescribed antimicrobial prophylaxis, and a follow-up was scheduled to begin chemotherapy with the goal of bone marrow transplant.
He was given four cycles of cyclophosphamide, hydroxydaunorubicin, oncovin, etoposide, prednisone (CHOEP) regimen with no treatment response. He switched to ifosfamide, carboplatin and etoposide (ICE) regimen, and received one cycle. Again, he switched treatment to dexamethasone, methotrexate, ifosfamide, L-asparaginase, and etoposide (SMILE) regimen and then nivolumab and brentuximab vedotin given the progressing disease. However, he did not respond to these therapies and passed away due to severe sepsis. Figure 15 depicts EBV titers throughout his entire disease course, and the effects of various chemotherapy regimens on his EBV titers. The rising EBV titers after chemotherapy depict resistance to treatment and refractory disease (Fig. 15). The patient was categorized as a high-risk patient with refractory disease, and the only curative option was to receive allogeneic hematopoietic stem cell transplant (allo-HSCT).
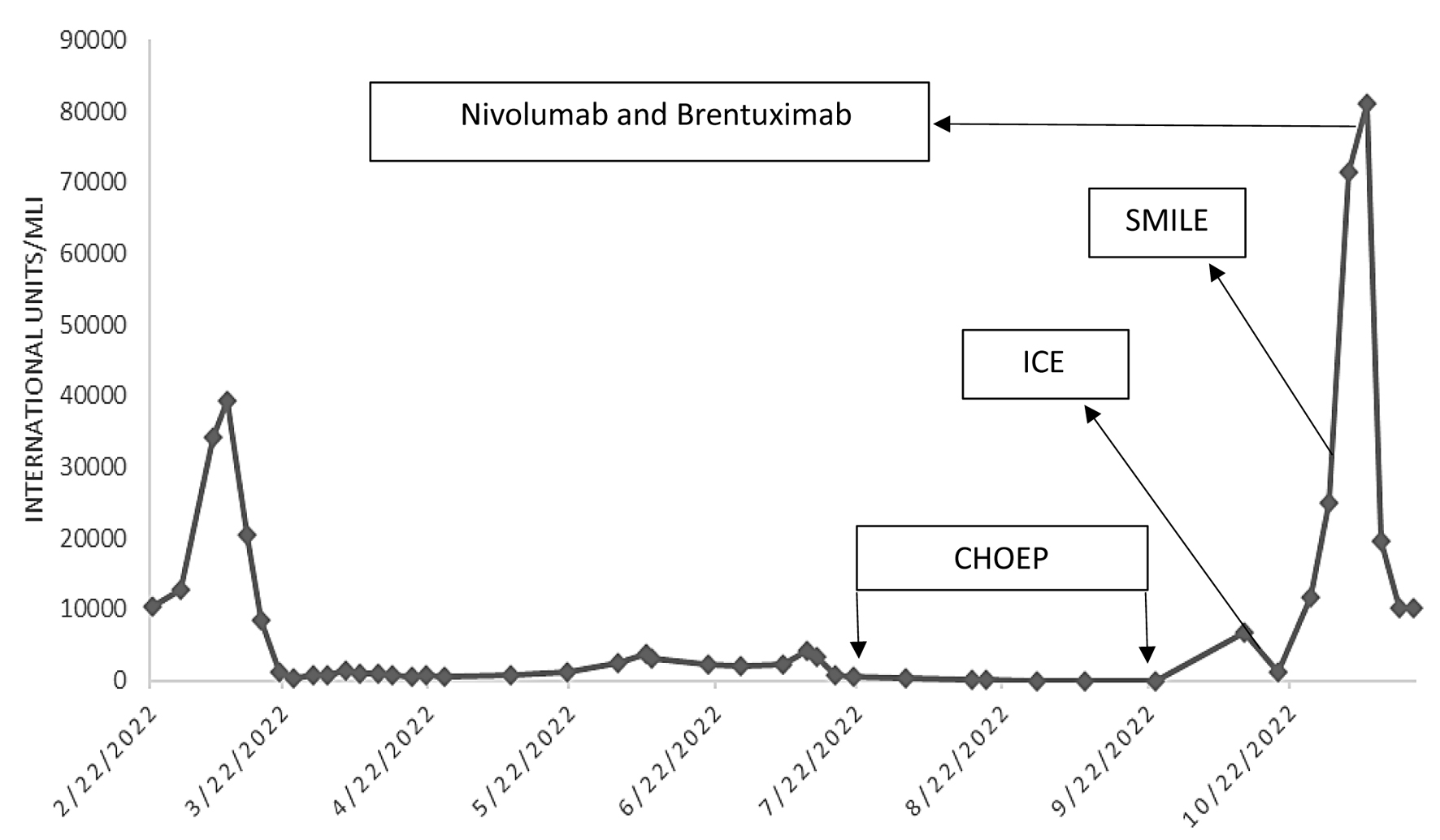 Click for large image | Figure 15. Epstein-Barr virus (EBV) DNA quantification via polymerase chain reaction. A line graph that charts EBV titers throughout the patient’s disease course, including the effects of chemotherapy. The arrows denote the start of chemotherapy regimen and the subsequent increase in titers indicates resistance to treatment. CHOEP: cyclophosphamide, hydroxydaunorubicin, oncovin, etoposide, prednisone; ICE: ifosfamide, carboplatin, and etoposide; SMILE: etoposide, ifosfamide, methotrexate and dexamethasone. |
| Discussion | ▴Top |
We report the first case, to our knowledge, of EBV+ TCL in a White patient who had a t(1;22)(p22;q11.2) translocation and underwent chemotherapy with the goal to induce complete remission (CR) and consolidate with allogenic stem cell transplant. Unfortunately, the patient passed away before this could be achieved.
The 2022 WHO classification of EBV+ T- and NK-cell lymphoid proliferations and lymphomas of childhood is subclassified into four major groups: severe mosquito bite allergy, hydroa vacciniforme lymphoproliferative disorder (LPD), systemic CAEBV, and systemic EBV+ TCL of childhood [3, 8]. Systemic EBV+ TCL of childhood is a rare disorder with a rapidly fatal disease course and poor prognosis. When associated with HLH, systemic EBV+ TCL presents with high fever, hepatosplenomegaly, pancytopenia, coagulopathy, and abnormal liver function tests. The median age of diagnosis is 20 years, and there is no correlation with the patient’s sex [9]. Patient mortality is high, with death often occurring only weeks or months after diagnosis [9]. Without high clinical suspicion, rapid diagnosis, and adequate treatment, death often occurs within weeks.
The treatment of systemic EBV+ TCL is nuanced in part because there are no consensus guidelines on the most appropriate first-line treatment. Evidence supporting any regimen is based on case reports and retrospective reviews. A common approach has been to induce remission with chemotherapy followed by consolidation and allogeneic stem cell transplant [10].
The choice of chemotherapeutic regimen, for systemic EBV+ TCL, typically includes etoposide and dexamethasone. Non-anthracycline-containing regimens that utilize etoposide or L-asparaginase have been efficacious against NK/T-cell lymphomas [11]. The chemotherapeutic agent etoposide has been proven in mice to have a high specificity against T-cell proliferation and cytokine secretion [11]. An observational study published in Blood in 2012 identified a cohort of 108 patients with EBV-associated T-cell or NK-cell LPDs, 53 of which were classified as systemic EBV+ T-cell LPD of childhood. Chemotherapy regimens in this study included CHOP, CHOP + etoposide, and cyclosporine A/etoposide/dexamethasone. However, only 7% of the patients that received chemotherapy in this study achieved a sustained CR [12]. We chose to treat our patient with CHOEP to induce remission, but his disease did not respond to treatment and was ultimately deemed refractory.
We chose to treat him with ICE regimen after his disease was refractory to CHOEP with the goal of pursuing an HSCT. In a study of 33 patients with extra nodal NK/T-cell lymphoma, chemotherapy regimens, such as (dexamethasone, etoposide, ifosfamide, carboplatin (DeVIC), combined with concurrent radiotherapy have shown an overall response rate (ORR) of 78% and a CR rate of 75% [13]. Our patient’s EBV titers continued to trend upward after one cycle of ICE. For relapsed or refractory NK/T-cell lymphomas that have not responded to anthracycline or etoposide-containing agents, salvage therapy with L-asparaginase-containing regimens has shown promise. L-asparaginase has been demonstrated to have excellent in vitro anti-NK/T-cell lymphoma activity [14[. In a phase 2 trial, SMILE therapy had an ORR of 80% and CR of 40% after two cycles [13]. We tried SMILE regimen but had no success in achieving remission, and his titers continued to trend upward.
Recently several new agents have become available for use in refractory peripheral T-cell lymphomas (PTCLs). Pralatrexate and romidepsin, for example, are approved broadly for PTCL with ORRs in large phase 2 studies of 29% and 25%, respectively [15, 16]. Brentuximab vedotin, a CD30-directed antibody-drug conjugate, has shown objective response in 41% of patients with relapsed TCLs including 54% of angioimmunoblastic T-cell lymphoma (AITL) patients [17]. A proportion of PTCLs (15-41% and 46-100%) overexpress programmed death ligand-1 (PD-L1) and CD30 [18, 19]. Thus, combination of Brentuximab vedotin and nivolumab could potentially be therapeutic. As per CheckMate 436 study, 33.3% of patients achieved CR and 12-month overall survival (OS) was 46.1% [20]. We, lastly, tried Brentuximab vedotin and nivolumab, which helped lower his EBV titers and control his disease. However, our patient passed away from sepsis before we could attempt an HSCT.
Allo-HSCT has been suggested as a potential cure for EBV+ TCL and could increase patient survival. While investigational data do not exist to support this approach for EBC+ TCL, specifically. In the same cohort of 108 patients, diagnosed with EBV-associated T-cell or NK-cell LPDs, studied by Kimura et al, 55% of the patients were given a stem cell transplant, with 60% of those with HLH achieving a CR [12]. OS was found to be statistically significant in this study, for T-cell LPD that received transplant (15-year OS: 56.9% vs. 36.1%; P = 0.003) [12]. Considering the complications and risks associated with allo-HSCT, it is typically reserved for high-risk patients with high-risk cytogenetics or patients with advanced relapsed/refractory disease.
There are active researches going on involving the role of novel therapies, including monoclonal antibodies, such as daratumumab, anti-PD-1 inhibitors, and JAK 1/2/3 inhibitors; however, these interventions have been primarily studied in extranodal NK/T-cell lymphomas [21]. Cytotoxic lymphocytes (CTLs) have also been examined targeting LMP1 and LMP2, which are weakly expressed on tumor cells of patients with NK-cell predominant TCL. The precise role of CTLs is not yet clear, as these have been studied mostly in the consolidative setting following radiotherapy and autologous stem cell transplant. Additionally, CTLs have not been studied in the diagnosis of systemic EBV+ T-cell LPD of childhood, and a major limitation in the use of CTLs is the timeline for manufacturing which typically takes weeks to months.
The phase 3 ALLELE trial was recently published and led to the approval of an off the shelf CTL, tabelecleucel, for relapsed/refractory EBV+ post-transplant lymphoproliferative disorder (PTLD) following solid organ transplant (SOT) or HSCT. In this trial, patients were required to have already received treatment with rituximab [22]. The objective response rates were promising with about 52% of HSCT recipients and 50% of SOT recipients achieving an objective response [22]. The treatment was also noted to be well tolerated, with the main toxicity being neutropenia [22]. Tabelecleucel specifically targets the EBV 3 latency profile to induce cytotoxic killing of all cells expressing the target antigens. This therapy has only been tested in EBV+ B-cell LPDs [22].
Our case illustrates that, while rare, this disease is not limited to Asian populations and can affect other ethnicities. Tabanelli et al found 14 cases of systemic EBV+ TCL of childhood among the European and US populations prior to 2011 [9]. Most cases were patients with Asian or Native American ethnic origins with only three cases among White patients [9].
The mutation seen in our patient, specifically the t(1;22)(p22;q11.2) mutation, warrants further investigation in patients with EBV+ TCL. An autopsy report from Japan investigating a 25-year-old man with DiGeorge syndrome who passed away from an EBV+ TCL noted a mosaic chromosome 22q11.2 deletion within the patient’s cardiac myocytes and lymphoma cells [23]. The 22q11.2 deletion has long been linked to DiGeorge syndrome, but the forementioned case report was the first reported association with an EBV+ T-cell malignancy [24]. Another report also found a constitutional t(1;22)(p22;q11.2) mutation in a 5-year-old patient with an ependymoma [25). While it is well known that this chromosome region is important for craniofacial development, the role it plays in thymus development and its association with rare immunologic cell derived malignancies may point towards an oncogenic mechanism [7]. The t(1;22)(p22;q11.2) breakpoint lies between the DiGeorge locus and the breakpoint cluster region, a significant component of the Philadelphia chromosome oncogene [7, 26]. A tumor suppressor gene may also exist within this region and is inactivated by additional systemic infections, such as EBV, leading to adverse outcomes. Additional research will clarify whether mutations in this chromosome region are causative or merely correlate with oncogenesis.
Conclusion
Our case report underscores the importance of retaining a wide differential diagnosis in an adult patient presenting with HLH, including unusual presentations of systemic EBV+ TCL of childhood, and highlights a possible new genetic locus associated with immunological malignancies. The case also highlights the need for more descriptive studies that clarify disease presentation and the role of genetics in disease progression. There is a notable lack of evidence on effective treatment strategies for systemic EBV+ TCL of childhood and role of stem cell transplant. We hope that future research in these areas will lead to the development of early detection methods and improved treatment options.
Acknowledgments
The authors thank Sarah Carey, MS, Jade Chang, and Jacalyn Newman, PhD, of Allegheny Health Network’s Health System Publication Support Office (HSPSO) for their assistance in editing and formatting the manuscript. The HSPSO is funded by Highmark Health (Pittsburgh, PA, USA), and all work was done in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
Financial Disclosure
The authors do not have any financial conflict of interest to disclose.
Conflict of Interest
None to declare.
Informed Consent
Informed consent was obtained by the patient’s family and can be provided from the corresponding author upon reasonable request.
Author Contributions
Lane Lerner, MS: original draft contributor; Sushanth Sreenivasan, MD: original draft contributor; Maitreyee Rai, MD: project management, draft review and editing; Chelsea Peterson, MD: draft review and final editing; Pragnan Kancharla, MD: draft review and editing. Mark Bunker, MD: image requisition and labeling; Samuel Santosa, MD: image requisition and labeling; Yazan Samhouri, MD: conceptualization, project management and review.
Data Availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
| References | ▴Top |
- Fox CP, Shannon-Lowe C, Gothard P, Kishore B, Neilson J, O'Connor N, Rowe M. Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in adults characterized by high viral genome load within circulating natural killer cells. Clin Infect Dis. 2010;51(1):66-69.
doi pubmed - Kasahara Y, Yachie A, Takei K, Kanegane C, Okada K, Ohta K, Seki H, et al. Differential cellular targets of Epstein-Barr virus (EBV) infection between acute EBV-associated hemophagocytic lymphohistiocytosis and chronic active EBV infection. Blood. 2001;98(6):1882-1888.
doi pubmed - Kim WY, Montes-Mojarro IA, Fend F, Quintanilla-Martinez L. Epstein-barr virus-associated T and NK-cell lymphoproliferative diseases. Front Pediatr. 2019;7:71.
doi pubmed pmc - Allen CE, McClain KL. Pathophysiology and epidemiology of hemophagocytic lymphohistiocytosis. Hematology Am Soc Hematol Educ Program. 2015;2015:177-182.
doi pubmed - Suzuki K, Ohshima K, Karube K, Suzumiya J, Ohga S, Ishihara S, Tamura K, et al. Clinicopathological states of Epstein-Barr virus-associated T/NK-cell lymphoproliferative disorders (severe chronic active EBV infection) of children and young adults. Int J Oncol. 2004;24(5):1165-1174.
pubmed - Stevens T, van der Werff Ten Bosch J, De Rademaeker M, Van Den Bogaert A, van den Akker M. Risk of malignancy in 22q11.2 deletion syndrome. Clin Case Rep. 2017;5(4):486-490.
doi pubmed pmc - Notarangelo LD. PIDs and cancer: an evolving story. Blood. 2010;116(8):1189-1190.
doi pubmed - Alaggio R, Amador C, Anagnostopoulos I, Attygalle AD, Araujo IBO, Berti E, Bhagat G, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia. 2022;36(7):1720-1748.
doi pubmed pmc - Tabanelli V, Agostinelli C, Sabattini E, Gazzola A, Bacci F, Capria S, Mannu C, et al. Systemic Epstein-Barr-virus-positive T cell lymphoproliferative childhood disease in a 22-year-old Caucasian man: A case report and review of the literature. J Med Case Rep. 2011;5:218.
doi pubmed pmc - Moskowitz AJ, Lunning MA, Horwitz SM. How I treat the peripheral T-cell lymphomas. Blood. 2014;123(17):2636-2644.
doi pubmed pmc - Johnson TS, Terrell CE, Millen SH, Katz JD, Hildeman DA, Jordan MB. Etoposide selectively ablates activated T cells to control the immunoregulatory disorder hemophagocytic lymphohistiocytosis. J Immunol. 2014;192(1):84-91.
doi pubmed pmc - Kimura H, Ito Y, Kawabe S, Gotoh K, Takahashi Y, Kojima S, Naoe T, et al. EBV-associated T/NK-cell lymphoproliferative diseases in nonimmunocompromised hosts: prospective analysis of 108 cases. Blood. 2012;119(3):673-686.
doi pubmed - Yamaguchi M, Tobinai K, Oguchi M, Ishizuka N, Kobayashi Y, Isobe Y, Ishizawa K, et al. Phase I/II study of concurrent chemoradiotherapy for localized nasal natural killer/T-cell lymphoma: Japan Clinical Oncology Group Study JCOG0211. J Clin Oncol. 2009;27(33):5594-5600.
doi pubmed - Ando M, Sugimoto K, Kitoh T, Sasaki M, Mukai K, Ando J, Egashira M, et al. Selective apoptosis of natural killer-cell tumours by l-asparaginase. Br J Haematol. 2005;130(6):860-868.
doi pubmed - Coiffier B, Pro B, Prince HM, Foss F, Sokol L, Greenwood M, Caballero D, et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol. 2012;30(6):631-636.
doi pubmed - O'Connor OA, Pro B, Pinter-Brown L, Bartlett N, Popplewell L, Coiffier B, Lechowicz MJ, et al. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol. 2011;29(9):1182-1189.
doi pubmed pmc - Horwitz SM, Advani RH, Bartlett NL, Jacobsen ED, Sharman JP, O'Connor OA, Siddiqi T, et al. Objective responses in relapsed T-cell lymphomas with single-agent brentuximab vedotin. Blood. 2014;123(20):3095-3100.
doi pubmed pmc - Wilcox RA, Feldman AL, Wada DA, Yang ZZ, Comfere NI, Dong H, Kwon ED, et al. B7-H1 (PD-L1, CD274) suppresses host immunity in T-cell lymphoproliferative disorders. Blood. 2009;114(10):2149-2158.
doi pubmed pmc - Bossard C, Dobay MP, Parrens M, Lamant L, Missiaglia E, Haioun C, Martin A, et al. Immunohistochemistry as a valuable tool to assess CD30 expression in peripheral T-cell lymphomas: high correlation with mRNA levels. Blood. 2014;124(19):2983-2986.
doi pubmed - Zinzani PL, Salles G, Moskowitz AJ, Santoro A, Mehta A, Barr PM, Mehta-Shah N, et al. Nivolumab plus brentuximab vedotin for relapsed/refractory peripheral T-cell lymphoma and cutaneous T-cell lymphoma. Blood Adv. 2024;8(10):2400-2404.
doi pubmed pmc - Lv K, Yin T, Yu M, Chen Z, Zhou Y, Li F. Treatment advances in EBV related lymphoproliferative diseases. Front Oncol. 2022;12:838817.
doi pubmed pmc - Nikiforow S, Whangbo JS, Reshef R, Tsai DE, Bunin N, Abu-Arja R, Mahadeo KM, et al. Tabelecleucel for EBV+ PTLD after allogeneic HCT or SOT in a multicenter expanded access protocol. Blood Adv. 2024;8(12):3001-3012.
doi pubmed pmc - Itoh S, Ohno T, Kakizaki S, Ichinohasama R. Epstein-Barr virus-positive T-cell lymphoma cells having chromosome 22q11.2 deletion: an autopsy report of DiGeorge syndrome. Hum Pathol. 2011;42(12):2037-2041.
doi pubmed - Morrow BE, McDonald-McGinn DM, Emanuel BS, Vermeesch JR, Scambler PJ. Molecular genetics of 22q11.2 deletion syndrome. Am J Med Genet A. 2018;176(10):2070-2081.
doi pubmed pmc - Park JP, Chaffee S, Noll WW, Rhodes CH. Constitutional de novo t(1;22)(p22;q11.2) and ependymoma. Cancer Genet Cytogenet. 1996;86(2):150-152.
doi pubmed - Laurent E, Talpaz M, Kantarjian H, Kurzrock R. The BCR gene and philadelphia chromosome-positive leukemogenesis. Cancer Res. 2001;61(6):2343-2355.
pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.