

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website http://www.thejh.org |
Case Report
Volume 3, Number 4, December 2014, pages 107-111
A Case of Langerhans Cell Histiocytosis With Risk Organ Involvement in a Young Adult
Audrey Kama, Brett Mahona, Reem Karmalib, c
aRush University Medical Center, Chicago, IL, USA
bSection of Hematology, Rush University Cancer Center, Chicago, IL, USA
cCorresponding Author: Reem Karmali, Section of Hematology, Rush University Cancer Center, 1725 W Harrison Street, Suite 809, Chicago, IL 60612, USA
Manuscript accepted for publication July 31, 2014
Short title: Langerhans Cell Histiocytosis
doi: http://dx.doi.org/10.14740/jh161w
| Abstract | ▴Top |
Multisystem Langerhans cell histiocytosis (MS-LCH) is a rare disease for which the standard of care has not been clearly established. We are the first to address frontline and salvage options for the management of an adolescent young adult (AYA) male with MS-LCH with “risk organ” involvement, a population for which there are no therapeutic guidelines. Our 33-year-old male patient presented with MS-LCH with generalized lymphadenopathy, hepatic and splenic involvement. He was initially treated with vinblastine and prednisolone with progression in disease. Our exploration of the literature revealed that salvage options in adults include single-agent chemotherapy, hematopoietic stem cell transplant, imatinib, and vemurafenib and are limited to small case series with questionable efficacy. We opted to use a pediatric regimen with the combination of cladribine and cytarabine and demonstrate that this approach can in fact be effective in an AYA patient.
Keywords: Langerhans cell histiocytosis; Multisystem Langerhans cell histiocytosis; Adolescent young adult; Cladribine; Cytarabine
| Introduction | ▴Top |
Langerhans cell histiocytosis (LCH) is a rare disease characterized by the proliferation and accumulation of dendritic cells, leading to organ dysfunction. It is most often diagnosed in childhood, but can occur at any age [1]. Morphologically, LCH cells stain positive for CD1a and/or CD207 (Langerin). Any organ/organ system can be affected; however, the most frequently affected include the skeleton, skin, pituitary, liver, spleen, hematopoietic system, lungs, lymph nodes and central nervous system (CNS) [2]. LCH is classified as either single system (SS-LCH) where one organ or system is involved, or multisystem (MS-LCH) where two or more systems are involved and may include “risk organs” (hematopoietic system, spleen and/or liver) [3]. Patients with SS-LCH are usually treated with local therapy, whereas MS-LCH requires systemic chemotherapy. Given the rarity of the disease, there is no clear standard of care, particularly data addressing the management of patients that fall within the relapsed population. There is even less data addressing patients that fall within the adolescent young adult (AYA) age group. We highlight an effective approach in such a patient.
| Case Report | ▴Top |
A 33-year-old male presented with abdominal pain and anemia with a hemoglobin of 9.6 g/dL. CT scans revealed diffuse lymphadenopathy, bilateral hydroureteronephrosis, attributed to retroperitoneal fibrosis and an infiltrative process around the tail of the pancreas, aorta, and kidney. Soon after, the patient was intubated for hypercapneic respiratory failure. An infectious workup was negative. An axillary lymph node biopsy revealed CD1a positive histiocytes, consistent with LCH (Fig. 1).
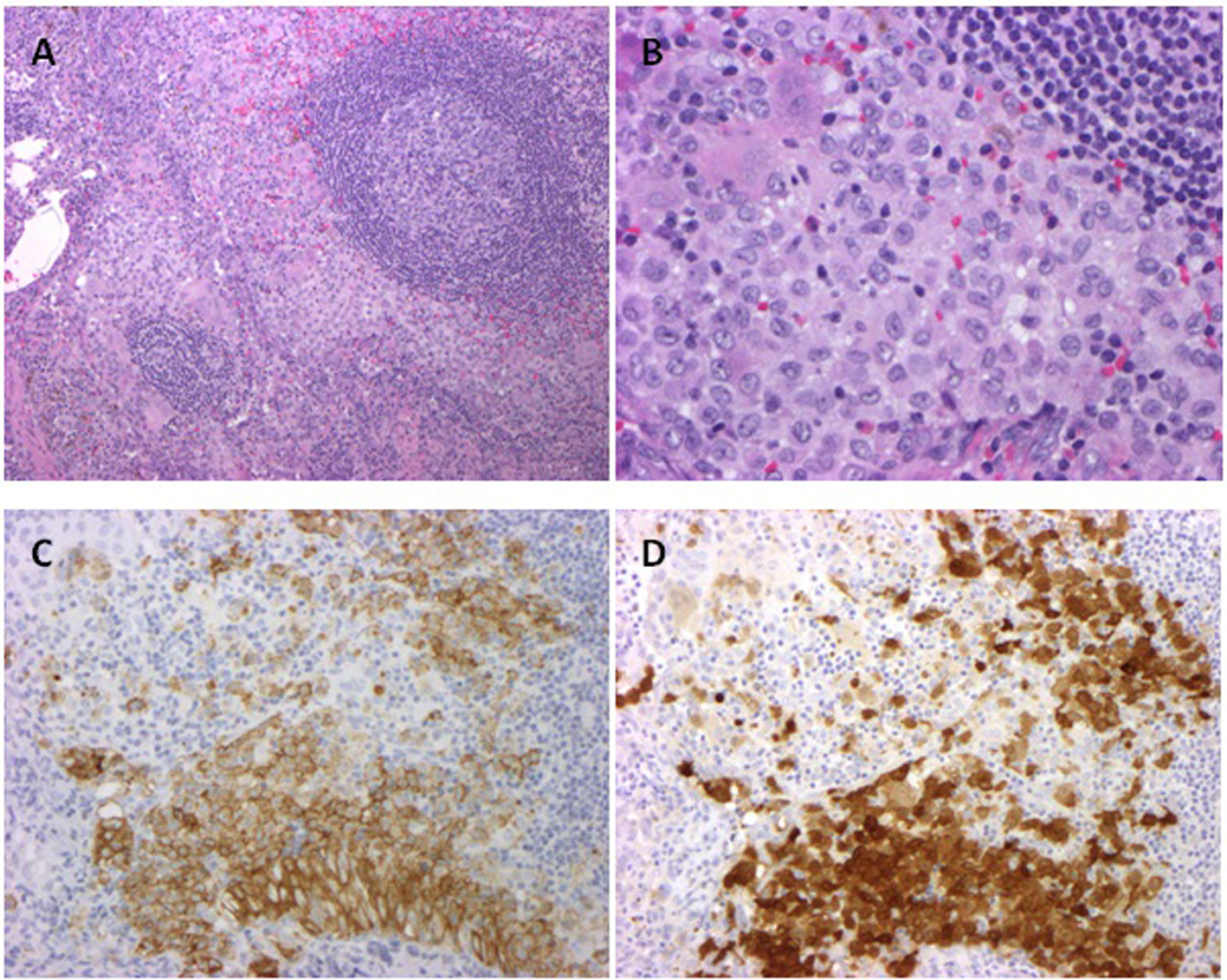 Figure 1. Axillary lymph node biopsy with Langerhans cell histiocytosis. A (× 100, hematoxylin and eosin). The lymph node shows paracortical and sinus Langerhans cells that spare the follicles; B. (× 400, hematoxylin and eosin): The Langerhans cells show irregularly grooved and folded nuclei. Admixed lymphocytes, eosinophils, plasma cells, and multinucleated giant cells are present; C. (× 200, CD1a): The LCH cells are strongly CD1a positive; D. (× 200, S100): The LCH cells are strongly S100 positive. LCH: Langerhans cell histiocytosis; CD 1a: cluster of differentiation 1a. Figure 1. Axillary lymph node biopsy with Langerhans cell histiocytosis. A (× 100, hematoxylin and eosin). The lymph node shows paracortical and sinus Langerhans cells that spare the follicles; B. (× 400, hematoxylin and eosin): The Langerhans cells show irregularly grooved and folded nuclei. Admixed lymphocytes, eosinophils, plasma cells, and multinucleated giant cells are present; C. (× 200, CD1a): The LCH cells are strongly CD1a positive; D. (× 200, S100): The LCH cells are strongly S100 positive. LCH: Langerhans cell histiocytosis; CD 1a: cluster of differentiation 1a. |
Staging revealed “risk organ” involvement: the patient had a transaminitis suggesting liver involvement, and splenomegaly. His bone marrow biopsy was negative for disease. He also had hypernatremia consistent with diabetes insipidus managed with DDAVP; an MRI of the brain incidentally showed hyperdense lesions around the optic nerves suggestive of LCH involvement (Fig. 2).
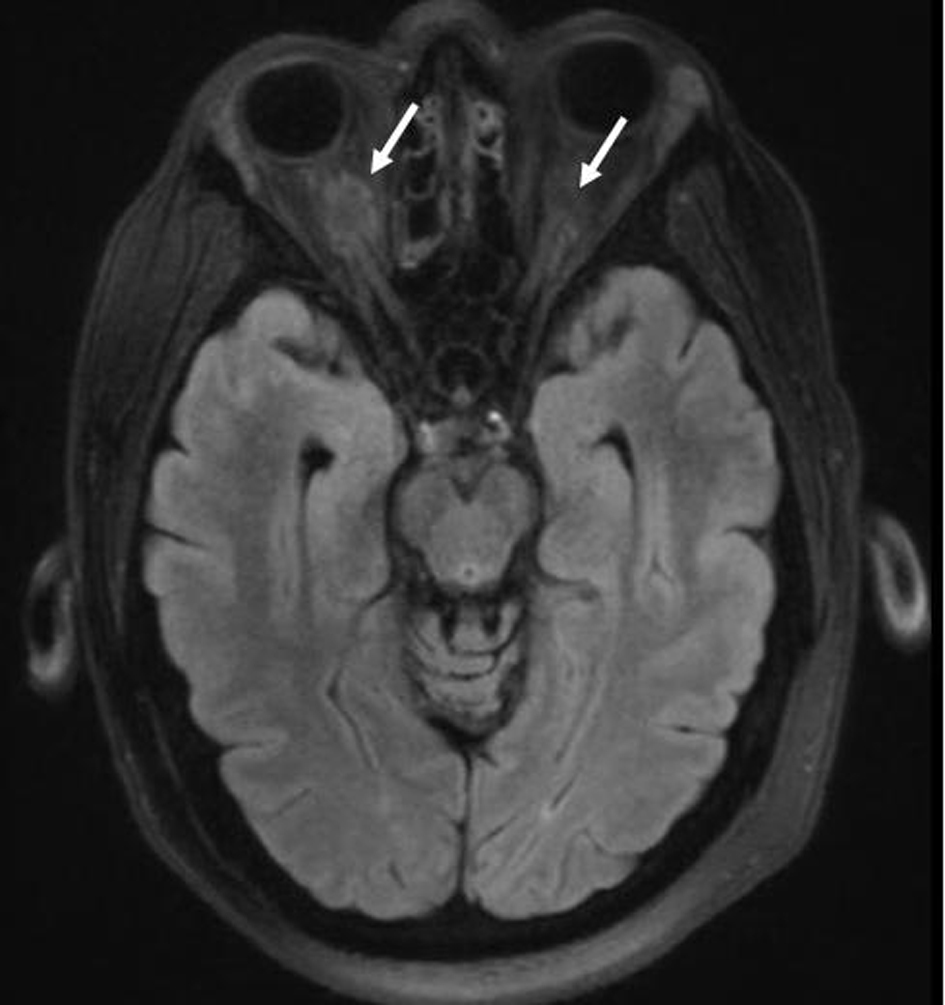 Figure 2. MRI of the brain demonstrated hyperdense lesions around the optic nerves, lesions appearing hypointense to the cerebral cortex on T2-weighted imaging, suggestive of Langerhans cell involvement. Figure 2. MRI of the brain demonstrated hyperdense lesions around the optic nerves, lesions appearing hypointense to the cerebral cortex on T2-weighted imaging, suggestive of Langerhans cell involvement. |
Therapy with prednisolone 125 mg daily and vinblastine weekly was initiated with resolution of respiratory failure within 1 week. Upon completion of this induction course, restaging scans demonstrated increasing axillary and inguinal adenopathy. B-RAF mutational analysis was negative. The patient was salvaged with cladribine and cytarabine for four cycles. Despite a 20% reduction in dose (given concern for toxicity), the patient’s course was complicated by prolonged cytopenias and sepsis. Restaging post-completion of therapy showed decrease of the soft tissue masses encasing the optic nerves, resolution of lymphadenopathy and stable mild splenomegaly. Surveillance scans 12 months later continue to demonstrate stability.
| Discussion | ▴Top |
The majority of frontline studies for MS-LCH have been directed at the pediatric population (Table 1) [4-7]. The Histiocyte Society conducted three study protocols (LCH-I, II, and III), the results collectively establishing vinblastine and prednisolone for 6 weeks in children as the current standard of therapy [1]. Conversely, in the adult population, there is no clear standard of treatment and most studies are limited by small sample size with some suggesting that vinblastine/prednisolone ± 6-mercaptopurine (6MCP) can be applied to the adult population with good response (Table 1) [8-14].
| Table 1. Summary of Studies for Management of MS-LCH in the Pediatric and Adult Populations |
In pediatric patients with refractory or relapsed MS-LCH, various therapeutic trials have been conducted as summarized in Table 2 [15-22]. Single agent cladribine has been studied with limited efficacy in patients with “risk organ” involvement [16, 17]. The combination of cladribine and cytarabine appears to achieve higher response rates with improved survival and low risk of reactivation [18, 19], serving as the basis for our treatment recommendations in our patient. More recently, higher survival rates have been demonstrated with clofarabine in cladribine refractory LCH [22].
| Table 2. Summary of Studies for Refractory/Relapsed MS-LCH in Pediatrics and Adults |
Literature addressing the management of refractory/relapsed MS-LCH in adults is much less robust, most consisting of case reports (Table 2) [12, 23-27]. More recently, B-RAF mutations have been observed in 38% to 69% of cases of LCH [27], making salvage with vemurafenib an appropriate choice in patients that harbor the mutation.
The current case report not only stresses the variability in the management of MS-LCH in pediatric and adult populations alike but also highlights another therapeutic challenge - whether patients in the AYA group should be treated with a pediatric or adult regimen. The frontline use vinblastine/prednisolone ± 6MCP (if “risk organs” are involved) appears reasonable in both children and adults [4-6, 8-10]. However, when faced with refractory/relapsed disease, there is no consensus on how to treat the AYA group. We demonstrate that the combination of cladribine and cytarabine, typically used in children, can in fact be effective in B-RAF negative AYA patients. That being said, toxicity with this regimen is significant supporting the need for further assessment of better tolerated and more effective therapies.
| References | ▴Top |
- Minkov M. Multisystem Langerhans cell histiocytosis in children: current treatment and future directions. Paediatr Drugs. 2011;13(2):75-86.
doi pubmed - Haupt R, Minkov M, Astigarraga I, Schafer E, Nanduri V, Jubran R, Egeler RM, et al. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013;60(2):175-184.
doi pubmed - Donadieu J, Chalard F, Jeziorski E. Medical management of langerhans cell histiocytosis from diagnosis to treatment. Expert Opin Pharmacother. 2012;13(9):1309-1322.
doi pubmed - Gadner H, Grois N, Arico M, Broadbent V, Ceci A, Jakobson A, Komp D, et al. A randomized trial of treatment for multisystem Langerhans' cell histiocytosis. J Pediatr. 2001;138(5):728-734.
doi pubmed - Gadner H, Grois N, Potschger U, Minkov M, Arico M, Braier J, Broadbent V, et al. Improved outcome in multisystem Langerhans cell histiocytosis is associated with therapy intensification. Blood. 2008;111(5):2556-2562.
doi pubmed - Gadner H, Minkov M, Grois N, Potschger U, Thiem E, Arico M, Astigarraga I, et al. Therapy prolongation improves outcome in multisystem Langerhans cell histiocytosis. Blood. 2013;121(25):5006-5014.
doi pubmed - Morimoto A, Ikushima S, Kinugawa N, Ishii E, Kohdera U, Sako M, Fujimoto J, et al. Improved outcome in the treatment of pediatric multifocal Langerhans cell histiocytosis: Results from the Japan Langerhans Cell Histiocytosis Study Group-96 protocol study. Cancer. 2006;107(3):613-619.
doi pubmed - Morimoto A, Shimazaki C, Takahashi S, Yoshikawa K, Nishimura R, Wakita H, Kobayashi Y, et al. Therapeutic outcome of multifocal Langerhans cell histiocytosis in adults treated with the Special C regimen formulated by the Japan LCH Study Group. Int J Hematol. 2013;97(1):103-108.
doi pubmed - von Stebut E, Schadmand-Fischer S, Brauninger W, Kreft A, Doberauer C, Steinbrink K. Successful treatment of adult multisystemic Langerhans cell histiocytosis with psoralen-UV-A, prednisolone, mercaptopurine, and vinblastine. Arch Dermatol. 2008;144(5):649-653.
doi pubmed - Matsuki E, Tsukada Y, Nakaya A, Yokoyama K, Okamoto S. Successful treatment of adult onset Langerhans cell histiocytosis with multi-drug combination therapy. Intern Med. 2011;50(8):909-914.
doi pubmed - Adam Z, Szturz P, Duras J, Pour L, Krejci M, Rehak Z, Koukalova R, et al. [Treatment of Langerhans cells histiocytosis by cladribin reached long-term complete remission in 9 out of 10 adult patients]. Klin Onkol. 2012;25(4):255-261.
pubmed - Pardanani A, Phyliky RL, Li CY, Tefferi A. 2-Chlorodeoxyadenosine therapy for disseminated Langerhans cell histiocytosis. Mayo Clin Proc. 2003;78(3):301-306.
doi pubmed - Derenzini E, Fina MP, Stefoni V, Pellegrini C, Venturini F, Broccoli A, Gandolfi L, et al. MACOP-B regimen in the treatment of adult Langerhans cell histiocytosis: experience on seven patients. Ann Oncol. 2010;21(6):1173-1178.
doi pubmed - Girschikofsky M, Arico M, Castillo D, Chu A, Doberauer C, Fichter J, Haroche J, et al. Management of adult patients with Langerhans cell histiocytosis: recommendations from an expert panel on behalf of Euro-Histio-Net. Orphanet J Rare Dis. 2013;8:72.
doi pubmed - Minkov M, Grois N, Broadbent V, Ceci A, Jakobson A, Ladisch S. Cyclosporine A therapy for multisystem langerhans cell histiocytosis. Med Pediatr Oncol. 1999;33(5):482-485.
doi - Weitzman S, Braier J, Donadieu J, Egeler RM, Grois N, Ladisch S, Potschger U, et al. 2'-Chlorodeoxyadenosine (2-CdA) as salvage therapy for Langerhans cell histiocytosis (LCH). results of the LCH-S-98 protocol of the Histiocyte Society. Pediatr Blood Cancer. 2009;53(7):1271-1276.
doi pubmed - Biswas G, Khadwal A, Arora B, Bhagwat R, Banavali SD, Nair CN, Pai SK, et al. Activity and toxicity of 2-CDA in Langerhans cell histiocytosis: a single institutional experience. Indian J Cancer. 2007;44(4):137-141.
doi pubmed - Bernard F, Thomas C, Bertrand Y, Munzer M, Landman Parker J, Ouache M, Colin VM, et al. Multi-centre pilot study of 2-chlorodeoxyadenosine and cytosine arabinoside combined chemotherapy in refractory Langerhans cell histiocytosis with haematological dysfunction. Eur J Cancer. 2005;41(17):2682-2689.
doi pubmed - Apollonsky N, Lipton JM. Treatment of refractory Langerhans cell histiocytosis (LCH) with a combination of 2-chlorodeoxyadenosine and cytosine arabinoside. J Pediatr Hematol Oncol. 2009;31(1):53-56.
doi pubmed - Steiner M, Matthes-Martin S, Attarbaschi A, Minkov M, Grois N, Unger E, Holter W, et al. Improved outcome of treatment-resistant high-risk Langerhans cell histiocytosis after allogeneic stem cell transplantation with reduced-intensity conditioning. Bone Marrow Transplant. 2005;36(3):215-225.
doi pubmed - Kudo K, Ohga S, Morimoto A, Ishida Y, Suzuki N, Hasegawa D, Nagatoshi Y, et al. Improved outcome of refractory Langerhans cell histiocytosis in children with hematopoietic stem cell transplantation in Japan. Bone Marrow Transplant. 2010;45(5):901-906.
doi pubmed - Simko SJ, Tran HD, Jones J, Bilgi M, Beaupin LK, Coulter D, Garrington T, et al. Clofarabine salvage therapy in refractory multifocal histiocytic disorders, including Langerhans cell histiocytosis, juvenile xanthogranuloma and Rosai-Dorfman disease. Pediatr Blood Cancer. 2014;61(3):479-487.
doi pubmed - Ingram W, Desai SR, Gibbs JS, Mufti G. Reduced-intensity conditioned allogeneic haematopoietic transplantation in an adult with Langerhans' cell histiocytosis and thrombocytopenia with absent radii. Bone Marrow Transplant. 2006;37(7):713-715.
doi pubmed - Ichikawa K, Nomura S, Ishii K, Okuno M, Kasai C, Maekawa T, Kadota E. Autologous stem cell transplantation for refractory Langerhans' cell histiocytosis. Bone Marrow Transplant. 2007;40(8):807-808.
doi pubmed - Konno S, Hizawa N, Betsuyaku T, Yasuo M, Yamamoto H, Koizumi T, Nishimura M. Adult Langerhans cell histiocytosis with independently relapsing lung and liver lesions that was successfully treated with etoposide. Intern Med. 2007;46(15):1231-1235.
doi pubmed - Janku F, Amin HM, Yang D, Garrido-Laguna I, Trent JC, Kurzrock R. Response of histiocytoses to imatinib mesylate: fire to ashes. J Clin Oncol. 2010;28(31):e633-636.
doi pubmed - Haroche J, Cohen-Aubart F, Emile JF, Arnaud L, Maksud P, Charlotte F, Cluzel P, et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood. 2013;121(9):1495-1500.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.