

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://www.thejh.org |
Case Report
Volume 10, Number 1, February 2021, pages 18-21
Delayed Hemolytic Transfusion Reaction in a Patient With Sickle Cell Disease and the Role of the Classical Complement Pathway: A Case Report
Pamela S. Haira, Timothy P. Hecka, Daniel T. Carra, Katherine D. Watsona, b, c, Jessica Pricea, d, Neel K. Krishnaa, e, Kenji M. Cunniona, b, c, e, f, William C. Owena, b, c
aDepartment of Pediatrics, Eastern Virginia Medical School, 700 West Olney Road, Norfolk, VA 23507, USA
bChildren’s Specialty Group, 811 Redgate Avenue, Norfolk, VA 23507, USA
cChildren’s Hospital of The King’s Daughters, 601 Children’s Lane, Norfolk, VA 23507, USA
dDepartment of Pharmacy, Children’s Hospital of The King’s Daughters, 601 Children’s Lane, Norfolk, VA 23507, USA
eDepartment of Microbiology and Molecular Cell Biology, Eastern Virginia Medical School, 700 West Olney Road, Norfolk, VA 23507-1696, USA
fCorresponding Author: Kenji M. Cunnion, Eastern Virginia Medical School, 700 West Olney Rd, Norfolk, VA 23508, USA
Manuscript submitted August 19, 2019, accepted October 6, 2020, published online February 6, 2021
Short title: DHTR - Ex Vivo Inhibition, PIC1
doi: https://doi.org/10.14740/jh553
| Abstract | ▴Top |
A 14-year-old female patient with sickle cell disease developed a severe delayed hemolytic transfusion reaction (DHTR) leading to multiple transfusions and intensive care management. To better understand the extent to which the classical complement pathway was contributing to her DHTR, we utilized the complement hemolysis using human erythrocytes (CHUHE) assay and the classical complement pathway inhibitor, PIC1. Residual discarded de-identified plasma and erythrocytes from the patient obtained from routine phlebotomy was acquired. These reagents were used in the CHUHE assay in the presence of increasing concentrations of PIC1. Complement-mediated hemolysis of the patient’s erythrocytes occurred in her plasma and complement permissive buffer. Increasing concentrations of PIC1 dose-dependently inhibited hemolysis to levels found for the negative control - complement inhibitor buffer. Complement-mediated hemolysis was demonstrated by the CHUHE assay for this patient with sickle cell disease and severe DHTR. PIC1 inhibition of hemolysis suggested that the classical complement pathway was contributing to her DHTR.
Keywords: Delayed hemolytic transfusion reaction; Complement; CHUHE; PIC1; Case report; Classical pathway
| Introduction | ▴Top |
A rare but life-threatening sequela of blood transfusion is delayed hemolytic transfusion reaction (DHTR). In patients with sickle cell disease (SCD), the clinical presentation of DHTR can be confused with vaso-occlusive crises (VOC) potentially delaying important interventions. It is believed that many DHTRs are mild and self-limited and thus frequently unidentified. However, a reduction in hemoglobin to the pre-transfusion level within 1 - 2 weeks post-transfusion is suspicious for DHTR [1]. Severe DHTR reactions can progress rapidly after the onset of non-localizing symptoms and are life-threatening. They are typically treated similarly to acute hemolytic reactions.
The mechanisms underlying DHTR remain poorly understood. The most commonly discussed theory is that DHTR occurs when a patient has been previously sensitized to an erythrocyte antigen, but has undetectable alloantibody levels at the time of transfusion [2]. One to four weeks after transfusion with erythrocytes bearing this antigen, a primary or anamnestic immune response may occur precipitating DHTR [3]. The antibody-coated donor erythrocytes, usually of the IgG subclass, are believed to be primarily destroyed by extravascular hemolysis in the liver and spleen via Fc-mediated phagocytosis [4]. A recent review notes that the risk of DHTR is substantially increased in persons with SCD with an incidence ranging from 1% to 20% of transfusions [5]. This review details the role of complement activation in acute hemolytic transfusions; however, the impacts of complement activation on DHTR remain largely unclear. A case series and a few case reports have described episodes of DHTR associated with direct antiglobulin test (DAT) positive for C3(d), sometimes in the setting of hyperhemolysis [6-9]. Another small case series and case reports have described the use of eculizumab for DHTR [10-12].
The complement hemolysis utilizing human erythrocytes (CHUHE) is an ex vivo assay that has been used to evaluate the contribution of antibody-initiated classical complement-mediated hemolysis in acute hemolytic transfusion reaction (AHTR) [13], ceftriaxone-induced immune hemolytic anemia (CIIHA) [14] and to assess hemolytic risk of blood type A plasmas [13]. PIC1, peptide inhibitor of complement C1, is a classical pathway complement inhibitor that blocks the enzymatic activity of the first component of the cascade, C1 [15]. PIC1 has previously been demonstrated to inhibit antibody-initiated complement-mediated hemolysis in models of acute hemolytic diseases like an ex vivo model of ABO incompatibility [16] and an in vivo model of AHTR [17]. Here we explored the potential ability of a complement inhibitor to moderate hemolysis ex vivo utilizing erythrocytes and plasma from a patient experiencing DHTR.
| Case Report | ▴Top |
Patient information and clinical findings
Our case describes a 14-year-old female patient with SCD (hemoglobin SS) who presented with VOC disease of the lower extremities. She then developed acute chest syndrome (ACS) requiring a packed red blood cell (RBC) transfusion. Seven days after the transfusion, she suffered acutely worsening bilateral thigh, arm and back pain. Her hemoglobin at this time was 7.6 g/dL with a reticulocyte count of 7.6%, compared with a post-transfusion hemoglobin of 8.8 g/dL, and a reticulocyte count of 8.8%. After admission to hospital, she developed an increasing oxygen requirement and her hemoglobin decreased to 5.0 g/dL overnight. She became febrile, hypertensive and developed respiratory distress with worsening non-localized pain.
Diagnosis
Her blood work showed decreasing platelets from 274,000 to 116,000/µL, a lactate dehydrogenase (LDH) of 5,317 U/L and a normal ADAMS13 level of 158%. Her DAT was negative and her urinalysis showed hemoglobinuria. Her hemoglobin S level was found to be rising (70.2%) compared with a value from 4 days prior (65.6%).
Treatment
A 500 mg pulse dose of methylprednisolone and 0.4 g/kg intravenous immunoglobulin (IVIG) infusion was given for suspicion of DHTR. She also received ceftriaxone and was transferred to the intensive care unit (ICU). During her first day in the ICU, her hemoglobin and platelet counts dropped to 4.1 g/dL and 50,000 cells/µL, respectively. She was then treated with eculizumab 900 mg, a 1 g infusion of rituximab, IV ferric carboxymaltose and 30,000 Units of erythropoietin alfa. Her hydroxyurea dose was held. She required escalating respiratory support to 100% FiO2 via 40 L high flow nasal cannula and exhibited acutely worsening perfusion on exam. Due to her worsening respiratory and clinical status, she was transfused with 5 mL/kg of packed RBCs. During day 2 in the ICU, her hemoglobin was 4.5 g/dL resulting in an additional transfusion with 5 mL/kg packed RBCs, which increased her hemoglobin to 5.3 g/dL. Her LDH was 25,071 U/L at that time. She also received a second dose of pulse IV methylprednisolone and 0.4g/kg IVIG. She began prophylactic enoxaparin with normal coagulation studies. Echocardiography showed elevated right ventricular pressure estimated at half systemic pressure. LDH peaked on ICU day 3 at 30,425 U/L and she was given a dose of 8 mg/kg tocilizumab and an IV methylprednisolone wean to 50 mg q6h was initiated. That evening, the hemoglobin decreased to 4.4 g/dL and her platelet count decreased. She experienced worsening respiratory failure requiring BiPap ventilation. She was transfused 5 mL/kg platelets and two additional 5 mL/kg aliquots of packed RBCs increasing her hemoglobin to 8.0 g/dL and platelet count to 107,000 cells/µL on ICU day 4. Subsequent echocardiogram demonstrated improved heart function.
By ICU day 5, the hemoglobin level and platelet counts remained stable and LDH began to trend downward. The patient was clinically improving and respiratory support was decreased. Antibiotics were discontinued after becoming afebrile and corticosteroids were further decreased. She was given another 900 mg dose of eculizumab on ICU day 8 and then transferred out of the ICU. At time of hospital discharge, her hemoglobin was 7.9 g/dL, her platelet count was normal, her LDH was 7,085 U/L and she was comfortable on room air.
Follow-up and outcomes
At follow-up 2 days later, she was asymptomatic and had a stable hemoglobin of 8.6 g/dL with a reticulocyte count of 18.2%. Eight days later, her hemoglobin dropped to 5.2 g/dL, but she was asymptomatic. She was readmitted to the hospital and treated with methylprednisolone, rituximab and eculizumab. She was subsequently discharged on a steroid taper and remained clinically and hematologically stable.
On our analysis, the patient’s plasma caused increased hemolysis of her erythrocytes in complement permissive buffer compared with complement inhibitory buffer (P = 0.059) in the CHUHE assay (Fig. 1). Addition of PIC1 showed a dose-dependent decrease of hemolysis that was statistically significant at the 2 mM (P = 0.036) and 4 mM dose (P = 0.029) compared with hemolysis in complement permissive buffer without PIC1. At higher doses PIC1 inhibited hemolysis to the background signal seen for complement inhibitory buffer (P > 0.21). These results demonstrate complement-mediated DHTR of this patient’s erythrocytes by her plasma that was inhibited in the presence of a classical pathway complement inhibitor.
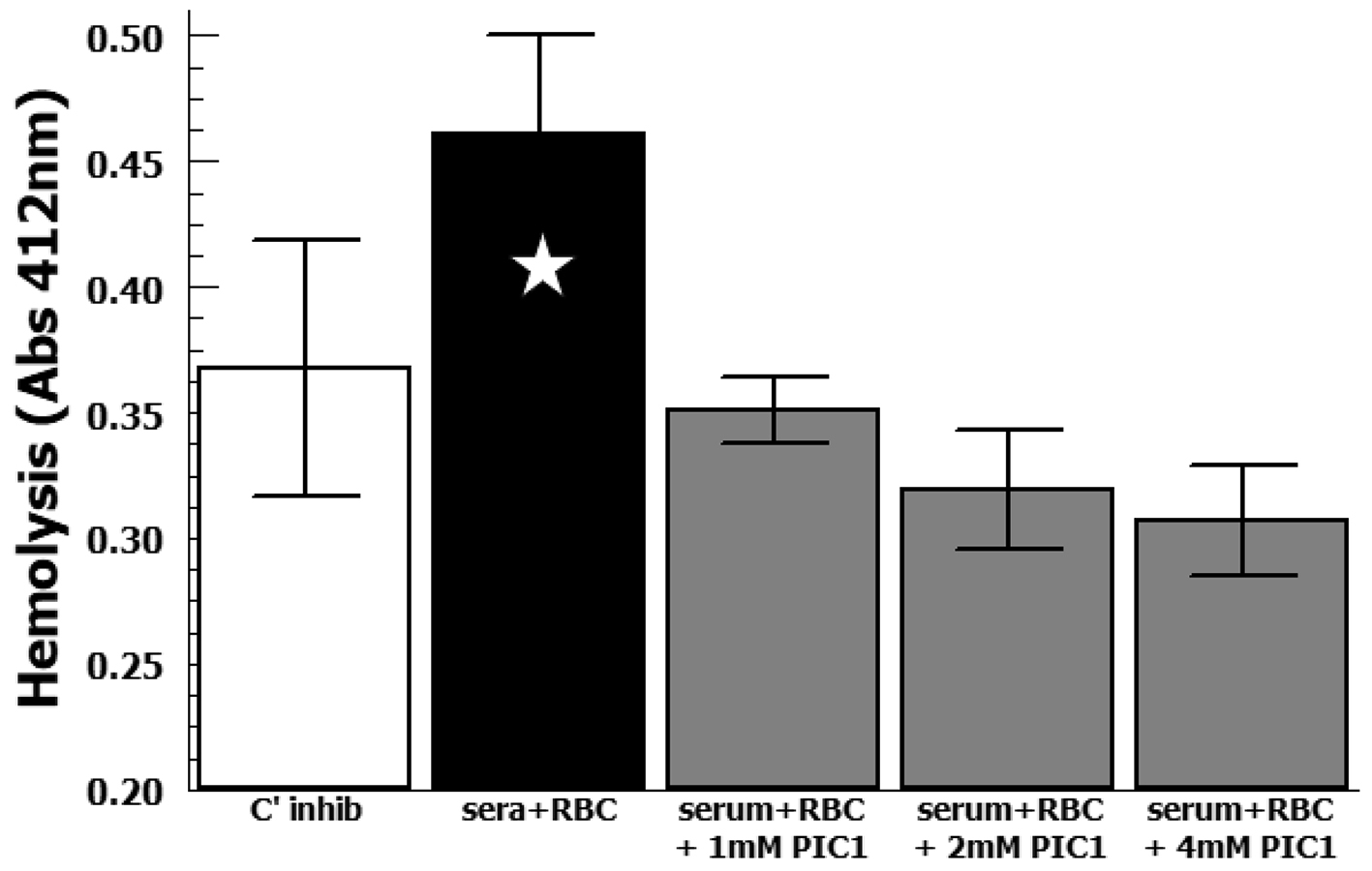 Click for large image | Figure 1. Analysis of hemolysis by CHUHE assay. DHTR subject’s erythrocytes were incubated with her plasma in complement inhibitory buffer (C’ inhib) or complement permissive buffer (sera + RBC). Increasing concentrations of PA-dPEG24 (PIC1) were added to the complement permissive reaction. Data (n = 3) are means of independent experiments ± SEM. Five-pointed star denotes P = 0.059. DHTR: delayed hemolytic transfusion reaction; CHUHE: complement hemolysis using human erythrocytes; RBC: red blood cell; SEM: standard error of mean. |
Ethics statement
This case report was reviewed by the Eastern Virginia Medical School IRB and determined to not constitute human subjects research. Blood and plasma were obtained as discarded de-identified samples from residual specimens in the Blood Bank.
| Discussion | ▴Top |
DHTR remains a challenging clinical diagnosis due to prolonged time between transfusion and hemolysis. For this patient with SCD and severe DHTR, complement-mediated hemolysis of her erythrocytes by her plasma was demonstrated ex vivo utilizing the CHUHE assay. It is unclear why her DAT was negative, although it is not uncommon to have a negative DAT in DHTR [18-20]. Unfortunately, this result led to a delay in recognizing that she was experiencing a DHTR and initiating interventions that could potentially have attenuated her disease process at an earlier stage. It is plausible that an assay like CHUHE could have allowed for more rapid identification of complement-mediated hemolysis contributing to her illness.
The classical complement pathway inhibitor PIC1 was able to inhibit her plasma lysing her erythrocytes ex vivo suggesting that antibody-initiated classical complement pathway activation can contribute to hemolysis in severe DHTR. The precipitous decline in this patient’s hemoglobin and clinical status are consistent with complement-mediated intravascular hemolysis contributing to her DHTR. Additional opsonization of erythrocytes with antibody as well as complement opsonins (e.g. C3b and iC3b) contribute to erythrocytes being removed from the blood by binding to FcR and complement receptors in the liver and spleen and subsequent lysis (i.e. extravascular hemolysis) [21]. This extravascular hemolysis occurring primarily in the reticuloendothelial system (RES) consists of Kupffer cells in the liver and macrophages in the spleen which are main parts of the reticuloendothelial system. The extent to which complement-mediated opsonization of her erythrocytes exacerbated extravascular hemolysis in the liver and spleen cannot be determined, but there is likely to be an additional contribution over antibody opsonization alone. These results raise the possibility that a classical complement pathway inhibitor could be used to moderate complement-mediated intravascular and extravascular hemolysis in a patient experiencing severe DHTR.
Learning points
DHTR can be a challenging diagnosis in patients with SCD and we have limited laboratory assays available currently to assist with patient care. Classical pathway of the complement system contributes to DHTR and this can be modified by a complement inhibitor to increase the lifespan of transfused RBCs.
Acknowledgments
None to declare.
Financial Disclosure
This work was funded in part by a grant from the Commonwealth Transfusion Foundation, Richmond, Virginia, USA, as well as NIH grant R21HL138710.
Conflict of Interest
Dr. Cunnion also serves as Chief Medical Officer for ReAlta Life Sciences, Inc. Dr. Krishna also serves as Chief Scientific Officer for ReAlta Life Sciences, Inc. The authors have no other potential conflict of interest to declare.
Informed Consent
Not applicable.
Author Contributions
All authors contributed substantively to retrieving and writing the case report or concept and design and execution of the experiments, or both. All authors contributed to drafting or editing the manuscript. All authors approve the final version of the manuscript. All authors agree to accountability to the accuracy and the integrity of the case and experimental work.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Strobel E. Hemolytic transfusion reactions. Transfus Med Hemother. 2008;35(5):346-353.
doi pubmed - Pirenne F, Yazdanbakhsh K. How I safely transfuse patients with sickle-cell disease and manage delayed hemolytic transfusion reactions. Blood. 2018;131(25):2773-2781.
doi pubmed - Delaney M, Wendel S, Bercovitz RS, Cid J, Cohn C, Dunbar NM, Apelseth TO, et al. Transfusion reactions: prevention, diagnosis, and treatment. Lancet. 2016;388(10061):2825-2836.
doi - Zimring JC, Spitalnik SL. Pathobiology of transfusion reactions. Annu Rev Pathol. 2015;10:83-110.
doi pubmed - Panch SR, Montemayor-Garcia C, Klein HG. Hemolytic Transfusion Reactions. N Engl J Med. 2019;381(2):150-162.
doi pubmed - Salama A, Mueller-Eckhardt C. Delayed hemolytic transfusion reactions. Evidence for complement activation involving allogeneic and autologous red cells. Transfusion. 1984;24(3):188-193.
doi pubmed - Greene DL, Khan S. Reactive lysis—a phenomenon of delayed hemolytic transfusion reactions. Immunohematology. 1993;9(3):74-77.
- Eberly LA, Osman D, Collins NP. Hyperhemolysis Syndrome without Underlying Hematologic Disease. Case Rep Hematol. 2015;2015:180526.
doi pubmed - Darabi K, Dzik S. Hyperhemolysis syndrome in anemia of chronic disease. Transfusion. 2005;45(12):1930-1933.
doi pubmed - Dumas G, Habibi A, Onimus T, Merle JC, Razazi K, Mekontso Dessap A, Galacteros F, et al. Eculizumab salvage therapy for delayed hemolysis transfusion reaction in sickle cell disease patients. Blood. 2016;127(8):1062-1064.
doi pubmed - Vlachaki E, Gavriilaki E, Kafantari K, Adamidou D, Tsitsikas D, Chasapopoulou E, Anagnostopoulos A, et al. Successful outcome of hyperhemolysis in sickle cell disease following multiple lines of treatment: the role of complement inhibition. Hemoglobin. 2018;42(5-6):339-341.
doi pubmed - Unnikrishnan A, Pelletier JPR, Bari S, Zumberg M, Shahmohamadi A, Spiess BD, Michael MJ, et al. Anti-N and anti-Do(a) immunoglobulin G alloantibody-mediated delayed hemolytic transfusion reaction with hyperhemolysis in sickle cell disease treated with eculizumab and HBOC-201: case report and review of the literature. Transfusion. 2019;59(6):1907-1910.
doi pubmed - Cunnion KM, Hair PS, Krishna NK, Whitley PH, Goldberg CL, Fadeyi EA, Maes LY. Discriminating complement-mediated acute transfusion reaction for type O+ red blood cells transfused into a B+ recipient with the complement hemolysis using human erythrocytes (CHUHE) assay. Transfusion. 2016;56(7):1845-1848.
doi pubmed - Cunnion KM, Feagin LM, Chicella MF, Kaszowski CL, Hair PS, Price J, Owen WC. Ceftriaxone-induced immune hemolytic anemia: in vitro reversal with peptide inhibitor of complement C1 (PIC1). Case Rep Hematol. 2019;2019:4105653.
doi pubmed - Sharp JA, Whitley PH, Cunnion KM, Krishna NK. Peptide inhibitor of complement c1, a novel suppressor of classical pathway activation: mechanistic studies and clinical potential. Front Immunol. 2014;5:406.
doi pubmed - Mauriello CT, Pallera HK, Sharp JA, Woltmann JL, Jr., Qian S, Hair PS, van der Pol P, et al. A novel peptide inhibitor of classical and lectin complement activation including ABO incompatibility. Mol Immunol. 2013;53(1-2):132-139.
doi pubmed - Kumar PS, Pallera HK, Hair PS, Rivera MG, Shah TA, Werner AL, Lattanzio FA, et al. Peptide inhibitor of complement C1 modulates acute intravascular hemolysis of mismatched red blood cells in rats. Transfusion. 2016;56(8):2133-2145.
doi pubmed - Talano JA, Hillery CA, Gottschall JL, Baylerian DM, Scott JP. Delayed hemolytic transfusion reaction/hyperhemolysis syndrome in children with sickle cell disease. Pediatrics. 2003;111(6 Pt 1):e661-665.
doi pubmed - Win N, Doughty H, Telfer P, Wild BJ, Pearson TC. Hyperhemolytic transfusion reaction in sickle cell disease. Transfusion. 2001;41(3):323-328.
doi pubmed - Petz LD, Calhoun L, Shulman IA, Johnson C, Herron RM. The sickle cell hemolytic transfusion reaction syndrome. Transfusion. 1997;37(4):382-392.
doi pubmed - Kato GJ, Gladwin MT. Mechanisms and clinical complications of hemolysis in sickle cell disease and thalassemia. In: Forget BG, Weatherall DJ, Higgs DR, Steinberg MH, editors. Disorders of hemoglobin: genetics, pathophysiology, and clinical management. 2 ed. Cambridge: Cambridge University Press; 2009. p. 201-224.
doi
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.