

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://www.thejh.org |
Case Report
Volume 11, Number 1, February 2022, pages 29-33

Compound Heterozygous Factor VII Deficiency c.1025G>A p.(Arg342Gln) With Novel Missense Variant c.194C>G p.(Ala65Gly)
Christian Aledia Gallardoa, b, c, f ![]() , Lester Jun Long Wonga, Christina Lai Lin Sumd, Liuh Ling Gohe, Kiat Hoe Onga, b, c, d, e
, Lester Jun Long Wonga, Christina Lai Lin Sumd, Liuh Ling Gohe, Kiat Hoe Onga, b, c, d, e
aDepartment of Haematology, Tan Tock Seng Hospital, Singapore, Singapore
bLee Kong Chian School of Medicine, Nanyang Technological University, Singapore, Singapore
cYong Loo Lin School of Medicine, National University of Singapore, Singapore, Singapore
dDepartment of Laboratory Medicine, Tan Tock Seng Hospital, Singapore, Singapore
eMolecular Diagnostic Laboratory, Tan Tock Seng Hospital, Singapore, Singapore
fCorresponding Author: Christian Aledia Gallardo, Department of Haematology, Tan Tock Seng Hospital, Singapore, Singapore
Manuscript submitted October 24, 2021, accepted January 4, 2022, published online February 26, 2022
Short title: Compound Heterozygous FVII Deficiency
doi: https://doi.org/10.14740/jh943
| Abstract | ▴Top |
Factor VII (FVII) deficiency manifests as prolonged prothrombin time (PT) and reduced FVII activity. We report a case of an asymptomatic 60-year-old gentleman with discrepancies in PT and FVII coagulant activity levels (FVII:C) on three different thromboplastin reagents used. Further sequence analysis on genomic DNA showed double heterozygosity for c.1025G>A p.Arg342Gln and c.194C>G p.Ala65Gly in the F7 gene. To date, p.Ala65Gly in exon 2 of the F7 gene represents a novel variant in patients with FVII deficiency and is classified as likely pathogenic. Computational prediction tools support a deleterious effect on the gene. The genotype-phenotype association and the clinical significance of this exon 2 missense variant is proposed in this case report.
Keywords: Factor VII deficiency; Prothrombin time; Thromboplastin; Mutation missense; Genetics
| Introduction | ▴Top |
Factor VII (FVII), also known as proconvertin, is a zymogen of vitamin K-dependent clotting protein that is synthesized by the liver [1]. Plasma levels range between 55% and 170%. It requires the presence of tissue factor (TF) to initiate coagulation [2]. Congenital or hereditary FVII deficiency manifests as prolonged prothrombin time (PT), normal activated partial thromboplastin time (APTT) and reduced FVII coagulant activity (FVII:C) by a one-stage PT-based assay. Congenital FVII deficiency is usually due to genetic variants in the F7 gene encoding FVII. The choice of thromboplastin may influence PT and FVII:C results [3].
FVII Padua is a variant form of FVII deficiency that was first described in 1978 [4]. It is a type II defect due to a (NM 019616.4) c1025G>A genetic variant in exon 8 of the F7 gene, resulting in a p.Arg342Gln alteration. This is characterized by a sharp discrepancy in FVII activity levels depending on the type of thromboplastins used [5, 6]. PT is prolonged when using rabbit brain thromboplastin, whereas assays using ox brain thromboplastin yield a normal result [7].
We present a patient with double heterozygous variants of F7 c.1025G>A p.Arg342Gln and in combination with a novel exon 2 missense variant c.194C>G p.Ala65Gly.
| Case Report | ▴Top |
The patient is a 60-year-old Chinese man, known to have hypertension, hyperlipidemia, and coronary artery disease. He had no bleeding tendencies and no prior surgeries. He presented with a fever for 10 days, diarrhea and loss of weight of 5 kg over 3 months. On arrival, he was febrile and was found to have non-tender hepatomegaly. The patient was not on any anti-platelet or anti-coagulant medication. Complete blood count revealed mild leukocytosis (12.5 × 109/L). Alanine aminotransferase and aspartate aminotransferase levels were elevated at 84 U/L and 46 U/L, respectively. PT was elevated (27.9 s) and APTT was within the normal range (34.3 s). His C-reactive protein was elevated (169.1 mg/L). Intravenous (IV) ceftriaxone and oral metronidazole were started. Computed tomography (CT) of his abdomen showed multiple hepatic abscesses, with the largest collection measuring 7.4 × 5.2 cm. The patient was initially planned for percutaneous aspiration and drainage of the liver abscess, but this was deferred in view of his coagulopathy.
A repeat CT scan of his abdomen after a prolonged course of IV antibiotics showed resolution of the hepatic abscesses. However, his PT was noted to be persistently elevated (27.6 - 29.9 s) despite the hepatic transaminases and other liver tests normalizing with the treatment of the hepatic abscesses. The PT test in our lab uses a rabbit brain thromboplastin reagent ((Neoplastine CI Plus, Stago Diagnostica). In view of this, Factors II, VII, and X activity levels and fibrinogen levels were measured. Fibrinogen was increased at 5g/L while the PT (Neoplastine CI Plus, Stago Diagnostica) was elevated. FVII activity level (FVII:C) (STA-Deficient VII, Stago Diagnostica) was low at 4%, while the other factor levels were normal (FII 121%, FX 130%). To further evaluate the low FVII:C activity, a total of 2 other thromboplastin reagents were used: (1) Recombinant human tissue factor and synthetic phospholipid (Dade Innovin, Siemens) and, (2) Lyophilized human placental thromboplastin (Thromborel S, Siemens). FVII:C levels using the recombinant human tissue factor and human placental thromboplastin assay were 28% and 21% respectively (Table 1). The VisuLize FVII ELISA Kit (Affinity Biologicals) quantified the FVII antigen level showed at 87%.
 Click to view | Table 1. Three Different Thromboplastin Reagents to Measure F7 Activity Levels |
In view of the discrepancy of FVII:C levels using the three thromboplastin reagents, sequence analysis was performed on genomic DNA extracted from peripheral blood using the QIAGEN® QIAamp® DNA Blood Mini Kit followed by polymerase chain reaction (PCR) amplification of the 5’ untranslated and coding regions in exons 1 to 8 of F7 (NG_009262.1) and bidirectional Sanger sequencing and capillary electrophoresis on the amplified products. Significance of genetic alterations were assessed based on prevalence, functional data as well as computational and predictive models. They were assigned a classification based on weighted evidence according to the American College of Medical Genetics recommendations [8]. The molecular analysis showed a double heterozygous state, with an exon 8 missense variant (NM_019616.4: c.1025G>A; p.Arg342Gln) in the active site domain and an exon 2 missense variant (NM_019616.4: c.194C>G; p.Ala65Gly) in the glutamic acid (GLA) domain (Fig. 1). The analysis could not determine the phasing of the two variants.
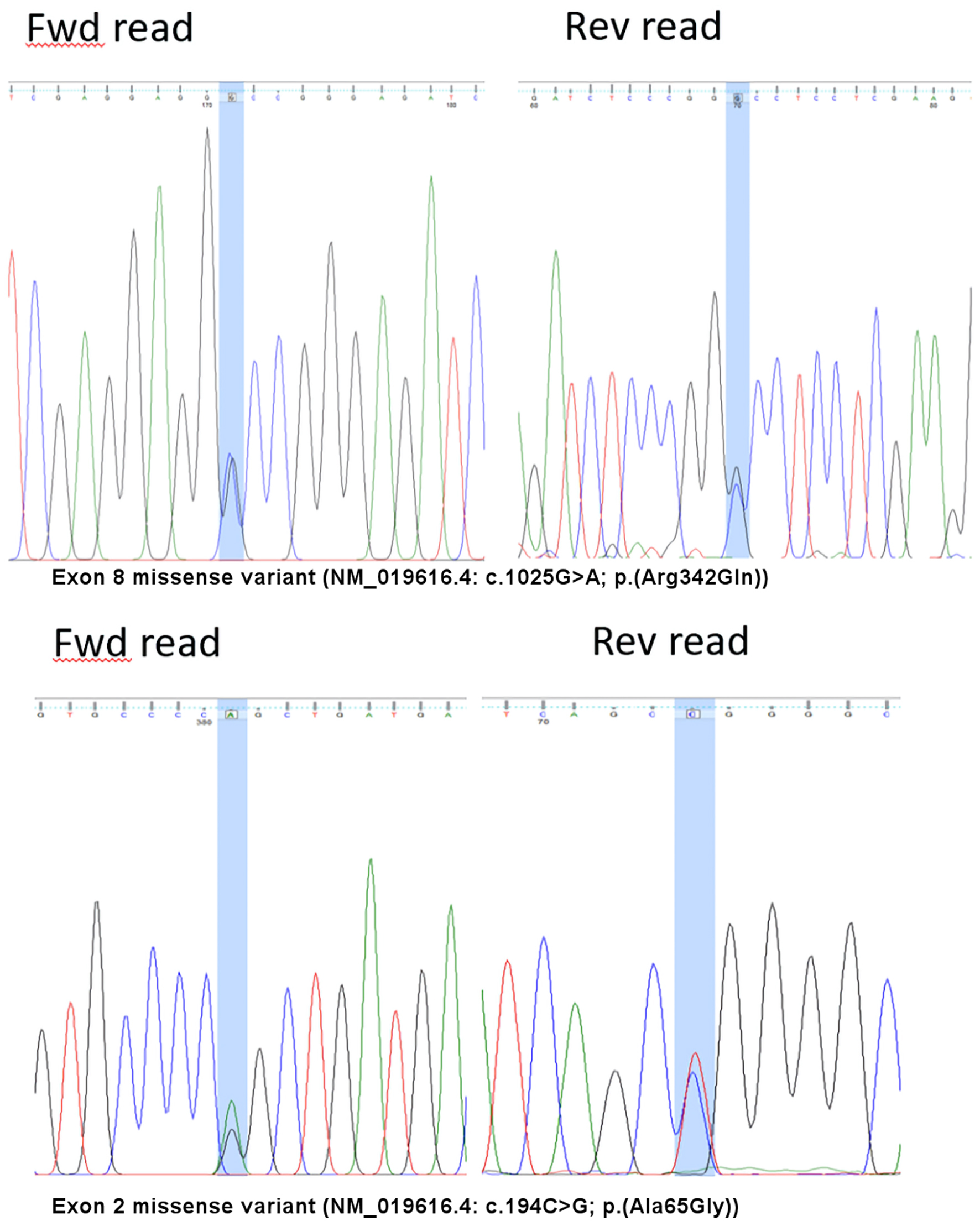 Click for large image | Figure 1. Variants detected on the F7 gene. Fwd: forward; Rev: reverse |
| Discussion | ▴Top |
It is known that inherited FVII deficiency has a wide spectrum of clinical phenotypes ranging from asymptomatic even in homozygous subjects to life-threatening bleeding [9]. The diagnosis of FVII deficiency is based on the presence of prolonged PT and confirmed by a one-stage PT-based assay demonstrating low FVII activity (FVII:C). In type I congenital FVII deficiency, FVII:C levels are low and comparable to antigen levels. In contrast, type II congenital FVII deficiency shows a discordant low FVII:C coupled with normal antigen levels [7]. The patient’s FVII antigen level was normal by an ELISA assay. FVII:C levels using three thromboplastin reagents demonstrated low levels, most marked with the rabbit brain thromboplastin and less so with the human tissue thromboplastin. It is unfortunate that ox brain thromboplastin was not available in Singapore. These differences are consistent with what was described by Girolami et al for FVII Padua [6, 7].
FVII:C levels are variable with wide ranges especially in patients with heterozygous mutations. In a review of 717 subjects from Europe and Latin America with variants in the F7 gene, it was found that patients with homozygous and compound heterozygous variants have a median FVII:C levels of 5% and 6%, respectively, with a combined range of < 1% to 31%. In comparison, patients with heterozygous mutations have a median FVII:C levels of 46% for asymptomatic patients and 39% in symptomatic patients, with a combined range of 12-67% [10]. With a FVII:C level of 4% using the rabbit brain thromboplastin, it is most likely that our patient had compound heterozygous alterations of the F7 gene.
The c.1025G>A variant in the F7 gene, which was seen in this patient, represents one of the most frequent missense variants in patients with FVII deficiency. It is sometimes reported using alternate nomenclature of pArg364Gln, and referred to as FVII Padua [11, 12]. FVII:C using human recombinant thromboplastin (28%) likely reflects the clinical phenotype more accurately as our patient was otherwise asymptomatic. This is also consistent with cases described in the literature where patients had either mild bleeding manifestations or were entirely asymptomatic [2].
To our knowledge, the c.194C>G; p.Ala65Gly variant is a novel variant in patients with FVII deficiency. According to the American College of Medical Genetics Guideline [13, 14], this variant p.Ala65Gly is classified as likely pathogenic (PM1, PM2, PP2, PP3, PP4). It represents a missense variant in exon 2 of the F7 gene encoding part of the GLA domain that is associated with calcium binding (PM1). Missense variants in F7 is a common mechanism of FVII deficiency (PP2). This reported variant is not found in the Genome Aggregation Database (GnomAD). It is also not listed in the F7 Gene Variant Database by the European Association of Haemophilia and Allied Disorders (EAHAD) [15]. In the Singapore SG10K Database, FVII p.Arg342Gln is rare (0.06% frequency in Chinese) while FVII p.Ala65Gly has no entry (PM2) [16]. In silico prediction tools, including MutationTaster, SIFT, FATHMN and Polyphen-2 that assess the functional impact of non-nonsynonymous variants based on a combination of sequence conservation and comparative genomics, unanimously support a deleterious effect on the gene (PP3) [17-19]. Lastly the alteration in FVII is highly specific for the patient’s phenotype (PP4).
Girolami et al reported [6] the FVII:C levels in a study of 6 homozygous and 14 heterozygous FVII Padua patients. Among the 6 homozygous FVII Padua population, the mean FVII:C activity levels using rabbit brain, human placenta or ox-brain thromboplastin were 8.8%, 33.3% and 100.2% respectively. Among the 14 heterozygous FVII Padua population, the mean FVII:C activity levels were 59.2%, 57.5% and 97.5% respectively using the same three thromboplastins. Our patient had FVII:C activity levels of 4%, 28%, 21% using rabbit brain, recombinant human tissue factor and human placental thromboplastins respectively. In view of discrepant FVII:C activity levels between our patient and the heterozygous FVII Padua population reported by Girolami et al [6], we postulate that the novel FVII p.Ala65Gly alteration also interferes with rabbit and human TF interaction with FVII.
It is known that FVII interacts extensively with soluble TF, with a binding interface that spans all domains [20]. As this TF binding interface likely includes the GLA domain, we postulate that the p.Ala65Gly alteration in our patient’s FVII further reduced its interaction with the various TF present in the different thromboplastin reagents used compared with a pure heterozygous FVII Padua patient. As the activation of the protease depends on the binding affinity with TF, we can also further postulate that the assay using human thromboplastin will most likely reflect the clinical bleeding tendency of this patient. This genotype-phenotype association and the clinical significance of the p.Ala65Gly alteration has yet to be confirmed. Further segregation and functional studies will be needed for the patient and his family members to understand the clinical significance of this alteration.
This case study illustrates the heterogeneous correlation of FVII levels with clinical bleeding tendency that is likely more frequently encountered in laboratories using nonhuman thromboplastin reagents. This is because as postulated above, the soluble TF in non-human thromboplastins likely has different binding affinity to human FVII than the soluble TF in human thromboplastin. Such laboratories should have access to another assay that uses human thromboplastin when encountering a low FVII level in a patient sample.
Acknowledgments
VisuLize FVII antigen kits were supported by Affinity Biologicals, ON, Canada.
Financial Disclosure
No funding from external source.
Conflict of Interest
The authors report no conflict of interest.
Informed Consent
Informed consent from the patient was obtained, and we have the consent for the molecular test as well.
Author Contributions
All authors made substantial contributions to the conception and writing the manuscript. CAG, CLLS, LJLW and KHO contributed to the literature search. CLLS performed laboratory investigation using different thromboplastin reagents. LLG helped in extracting the genomic DNA sequencing.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Mariani G, Bernardi F. Factor VII deficiency. Semin Thromb Hemost. 2009;35(4):400-406.
doi pubmed - Sevenet PO, Kaczor DA, Depasse F. Factor VII deficiency: from basics to clinical laboratory diagnosis and patient management. Clin Appl Thromb Hemost. 2017;23(7):703-710.
doi pubmed - Bolton-Maggs PH, Hay CR, Shanks D, Mitchell MJ, McVey JH. The importance of tissue factor source in the management of Factor VII deficiency. Thromb Haemost. 2007;97(1):151-152.
doi pubmed - Girolami A, Fabris F, Dal Bo Zanon R, Ghiotto G, Burul A. Factor VII Padua: a congenital coagulation disorder due to an abnormal factor VII with a peculiar activation pattern. J Lab Clin Med. 1978;91(3):387-395.
- O'Brien DP, Gale KM, Anderson JS, McVey JH, Miller GJ, Meade TW, Tuddenham EG. Purification and characterization of factor VII 304-Gln: a variant molecule with reduced activity isolated from a clinically unaffected male. Blood. 1991;78(1):132-140.
doi pubmed - Girolami A, Bertozzi I, de Marinis GB, Bonamigo E, Fabris F. Activated FVII levels in factor VII Padua (Arg304Gln) coagulation disorder and in true factor VII deficiency: a study in homozygotes and heterozygotes. Hematology. 2011;16(5):308-312.
doi pubmed - Girolami A, Berti de Marinis G, Bonamigo E, Sartori R, Vettore S. Ox brain versus rabbit brain thromboplastin assays are the best tool for a preliminary diagnosis of the Arg304Gln factor VII defect (FVII Padua). Acta Haematol. 2010;124(4):229-234.
doi pubmed - Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, Tsimberidou AM, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. 2017;19(1):4-23.
doi pubmed - Napolitano M, Siragusa S, Mariani G. Factor VII deficiency: clinical phenotype, genotype and therapy. J Clin Med. 2017;6(4):38.
doi pubmed - Herrmann FH, Wulff K, Auerswald G, Schulman S, Astermark J, Batorova A, Kreuz W, et al. Factor VII deficiency: clinical manifestation of 717 subjects from Europe and Latin America with mutations in the factor 7 gene. Haemophilia. 2009;15(1):267-280.
doi pubmed - Antonio G, Mirta A, Emanuel S, Graciela S, Silvia F, Maria LA, et al. First report of homozygous factor VII Padua (Arg304Gln) defect in a family from Argentina. Res Artic Hematol Med Oncol Hematol Med Oncol. 2016;1(1):1-5.
doi - Rabelo FY, Jardim LL, Landau MB, Gadelha T, Correa MF, Pereira IF, Rezende SM. The molecular basis of low activity levels of coagulation factor VII: a Brazilian cohort. Haemophilia. 2015;21(5):670-680.
doi pubmed - VarSome [Internet]. Available from: https://varsome.com/variant/hg19/F7 Ala87Gly.
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-424.
doi pubmed - Factor VII variant database [Internet]. Available from: https://f7-db.eahad.org/.
- Wu D, Dou J, Chai X, Bellis C, Wilm A, Shih CC, Soon WWJ, et al. Large-scale whole-genome sequencing of three diverse Asian populations in Singapore. Cell. 2019;179(3):736-749.e715.
- Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012;40(Web Server issue):W452-W457.
doi pubmed - Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GL, Edwards KJ, Day IN, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat. 2013;34(1):57-65.
doi pubmed - Quang D, Chen Y, Xie X. DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics. 2015;31(5):761-763.
doi pubmed - Gajsiewicz JM, Morrissey JH. Structure-function relationship of the interaction between tissue factor and factor VIIa. Semin Thromb Hemost. 2015;41(7):682-690.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.