

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://www.thejh.org |
Original Article
Volume 11, Number 1, February 2022, pages 8-14

Hematopoietic Stem Cell Transplantation Stabilizes Cerebral Vasculopathy in High-Risk Pediatric Sickle Cell Disease Patients: Evidence From a Referral Transplant Center
Abdullah Al-Jefria, b, Khawar Siddiquia, Amira Al-Oraibia, Amal Al-Seraihya, Ali Al Ahmaria, Ibrahim Ghemlasa, Awatif Al Anazia, Hawazen Al Saedia, Mouhab Ayasa
aDepartment of Pediatric Hematology/Oncology, King Faisal Specialist Hospital and Research Center, Riyadh, Saudi Arabia
bCorresponding Author: Abdullah Al-Jefri, Department of Pediatric Hematology/Oncology, King Faisal Specialist Hospital and Research Center, Riyadh 11211, Saudi Arabia
Manuscript submitted November 4, 2021, accepted December 28, 2021, published online February 26, 2022
Short title: HSCT in Pediatric SCD With CV
doi: https://doi.org/10.14740/jh949
| Abstract | ▴Top |
Background: Severe sickle cell disease (SCD) can present with different vaso-occlusive manifestations with cerebral vasculopathy (CV) as one of the most serious complications. Hematopoietic stem cell transplant (HSCT) is the ultimate therapy for this complication. The aim of this study was to assess the outcome and impact of HSCT on severe SCD patients with CV complications.
Methods: Twenty-five consecutive transplants-naive pediatric SCD patients with CV complications underwent HSCT at our institution between 1993 and 2015, using bone marrow as stem cells source from fully match related donors were included. Neurologic evaluation was done both clinically and radiologically before transplantation and regularly following the HSCT.
Results: With a median follow-up of 52.2 ± 5.8 months, the cumulative probability of overall survival (OS) at 3 years was 92.0% and event-free survival (EFS) was 88%. Significant neurologic improvements were observed in most of the patients clinically. Different neurologic complications were assessed. The neurologic manifestations before and after HSCT were hemiparesis (11, 1), seizures (13, 8), focal neurologic deficit (4, 2), loss of conscious (2, 1) headache (6, 1), and psychological symptoms (5, 2). Post-HSCT radiological imaging was done in 15 patients, which showed stabilization of CV among all.
Conclusions: Allogeneic HSCT in patients with severe SCD presenting with CV complications including moyamoya vasculopathy showed favorable outcome with significant clinical neurologic improvement and stabilization of the disease. None of the patients with severe vasculopathy underwent neurological vascular by-pass surgery prior to HSCT.
Keywords: Stem cell transplantation; Pediatric sickle cell disease; Cerebral vasculopathy; Overt stroke; Neurologic deficit; Convulsions
| Introduction | ▴Top |
Sickle cell disease (SCD) is a major health challenge in Saudi Arabia. The prevalence of SCD in Saudi Arabia varies significantly in different parts of the country, with the highest prevalence in the Eastern Province, followed by the Southwestern Province. The reported prevalence for sickle-cell trait ranges from 2% to 27% [1-4]. Overt stroke occurs in 7-13% of children with SCD and can lead to motor disability, neuropsychological impairment and death [5, 6]. Reports on the incidence of stroke in SCD in Saudi Arabia are limited. From a hospital-based study of SCD in the western part of Saudi Arabia, the incidence of overt stroke in children was reported to be 9.4% in a predominantly African (severe) haplotype [7-9]. While in Eastern Province this statistic is around 6% [10]. It is expected to be lower considering its less severe phenotype due to the high hemoglobin F percentage (Arab-Indian haplotype) compared to the severe African type seen in the Southwestern region, which all of our patient cohort represents [7]. SCD patients can present with serious complications, such as cerebral vasculopathy (CV), leading to stroke and other neurologic deficit [6, 8, 11, 12]. Such clinical manifestation can be elicited by magnetic resonance imaging/angiogram (MRI/MRA) that can show spectrum of radiological abnormalities indicative of infarction, ischemia and atrophy (Fig. 1) [13, 14].
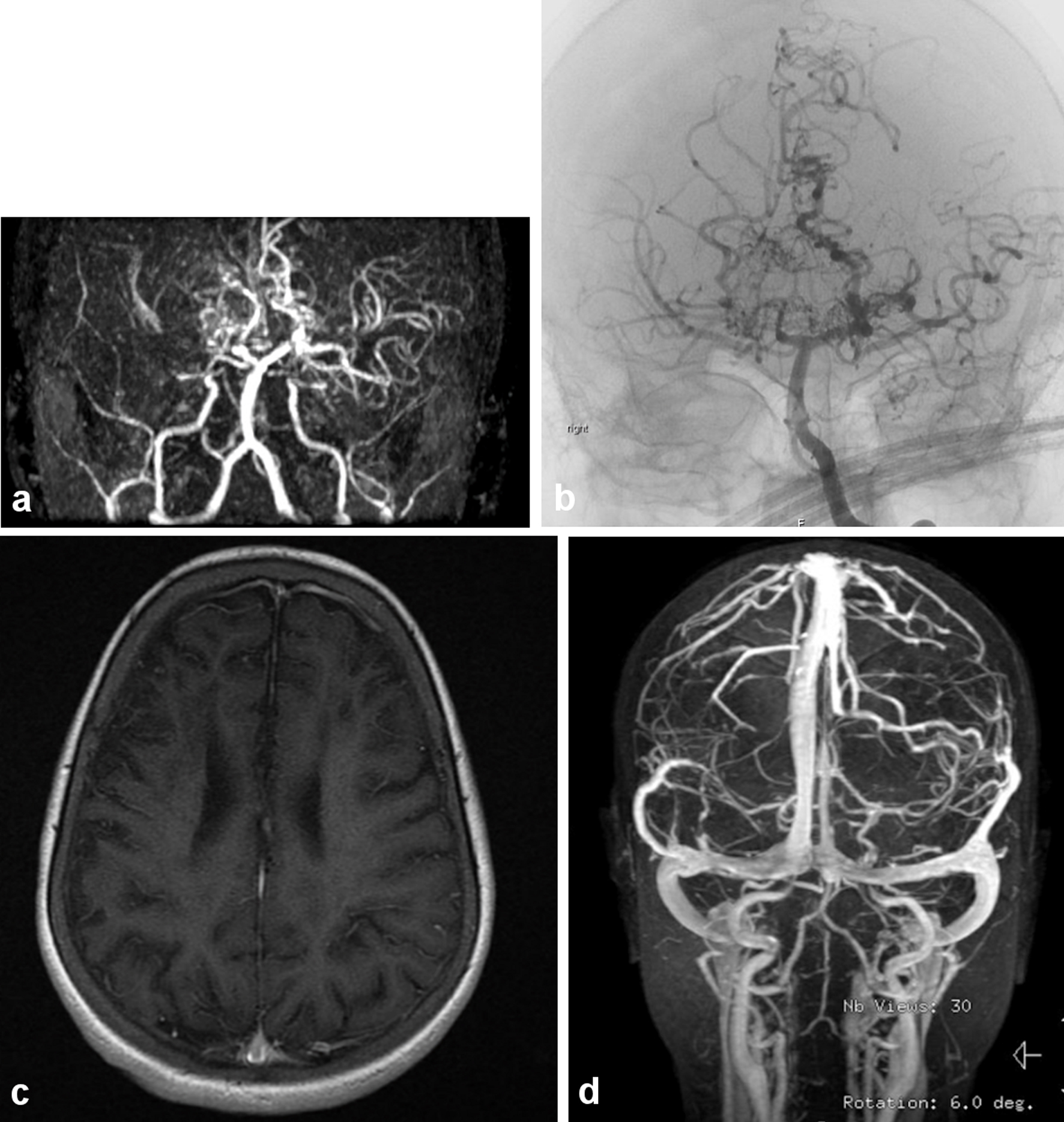 Click for large image | Figure 1. (a) The magnetic resonance (MR) angiography of the intracranial circulation showed significant prominent collateral vessels in the posterior cerebral artery territory with evidence for probable retrograde filling of middle cerebral artery territory from the posterior circulation. Significant collateral vessel prominence especially in the perforator branches in the thalamic region is detected. The findings are consistent with moyamoya disease with major thinning and attenuation in the supraclinoid internal carotid artery, and associated multiple watershed zone infarctions (before HSCT: March, 2009). (b) Evidence of narrowing at the distal internal carotid artery in the supraophthalmic segment bilaterally with the development of significant amount of small perforators giving puff of smoke appearance characteristic of moyamoya disease (same patient after HSCT in July, 2011). (c) Significant cortical and subcortical encephalomalacia diffusely along supratentorial brain parenchyma, significant within right frontal lobe with evidence of hemodynamic ischemic changes along the bilateral centrum semiovale. (d) Magnetic resonance imaging (MRI) with normal vascular distribution with moderately severe sickle cell disease. |
About 5% to 17% of SCD patients can develop stroke during childhood and adolescence [15]. The recurrence rate of infarction without regular monthly transfusion in patients who have had a prior clinical stroke can be as high as 67% [16, 17]. Hematopoietic stem cell transplant (HSCT) had shown a remarkable potential cure for SCD [18-21]. For patients with CV, studies have shown improvement in neurological deficit as well as vascular improvement after HSCT [20, 22-29]. We conducted an observational study to review and describe the outcome of HSCT in pediatric SCD patients presenting with CV at our institution.
| Materials and Methods | ▴Top |
Study design
From January, 1993 to December, 2015, 36 pediatric SCD patients (age ≤ 14 years) were referred to us for possible HSCT. Twenty-five patients had full matched human leukocyte antigens (HLA) identical related donors and underwent HSCT, which are the focus of this study. This is a retrospective account of their transplant outcome. Data on clinical, radiologic, transplant-related parameters including donor HLA type, post-transplant chimerism, engraftment, incidence of graft-versus-host disease (GVHD), infectious and non-infectious toxicity and outcome were downloaded from institutional databases, being populated prospectively. All parents and/or guardians signed institutional review board-approved parental informed consent prior to the transplant.
Patients
All patients were transplant-naive and were evaluated clinically for CV. MRI/MRA was the radiological method of evaluating patients with neurologic abnormality. None of the patients had undergone cerebral re-vascularization surgery like encephaloduroarteriosynangiosis (EDAS) or encephalomyoarteriosynangiosis (EMAS) prior to the transplant [30]. All recipients had normal renal and cardiac function at the time of transplant.
Transplant procedure
Patients were conditioned with myeloablative regimen, which consisted of busulfan, cyclophosphamide and anti-thymocyte globulin (ATG) in 19 (76.0%) patients, while ATG was not a part of the conditioning regimen for the remaining six (24.0%) cases. GVHD prophylaxis was in the form of short methotrexate and cyclosporine. Bone marrow was the source of stem cells in all cases.
Patients were hospitalized in single rooms with high-efficiency particulate air filtration with positive pressure until neutrophil recovery. Patients received acyclovir prophylaxis if they were seropositive for herpes simplex virus and/or cytomegalovirus (CMV). Oral trimethoprim-sulfamethoxazole was given for Pneumocystis prophylaxis after engraftment for 1 year. Broad-spectrum intravenous (IV) antibacterial and antifungal and/or antiviral antimicrobials were administered for fevers as indicated. All patients received granulocyte colony-stimulating factor 5 µg/kg per day subcutaneously, from the day after transplant until neutrophil recovery. CMV reactivation was monitored weekly until at least day 100 following transplant and preemptively treated with ganciclovir or foscarnet. No prophylactic rituximab was given.
End point definitions
Time to neutrophil recovery was the first of 3 consecutive days on which the absolute neutrophil count (ANC) was ≥ 0.5 × 109/L. Primary graft failure was defined as failure to achieve an ANC of 0.5 × 109/L by day 42, and secondary graft failure as an ANC < 0.5 × 109/L for 3 consecutive days, or 0% donor chimerism by polymerase chain reaction (PCR) in patients who have achieved an ANC of ≥ 0.5 × 109/L. Time to platelet recovery was the first of 3 consecutive days on which the platelet count was > 20 × 109/L without transfusions for 7 days before the first measurement. GVHD was graded by standard criteria. Graft failure and death from all causes was considered as an event. Overall survival (OS) was taken from infusion to the expiry or the last contact date, while event-free survival (EFS: probability of survival with sustained donor cell engraftment) was taken from the day of infusion till the disease recurrence date, with death from any cause or graft failure as an event, whichever came first.
Statistical considerations
All continuous data were presented as median with minimum and maximum points. Kaplan-Meier curves were drawn for survival analyses. McNemar’s test was used to test for the independence of differences for the number of patients for observed symptoms in the group. IBM-SPSS for Windows (version 20.0) was used for statistical analysis of the data.
Ethical considerations
This study was submitted to the Institutional Review Board of King Faisal Specialist Hospital and Research Center, Riyadh, Saudi Arabia before initiation, and was approved by the Research Advisory Committee through established procedures via approval number 2171164; and the study was conducted in compliance with the ethical standards of the responsible institution on human subjects as well as with the Helsinki Declaration.
| Results | ▴Top |
A total of 25 pediatric patients (age ≤ 14 years) with SCD underwent consecutive HSCT from January 1993 to December 2015 at our institution were included. Median age at HSCT was 10.6 years (range: 3.0 - 13.9 years); 20 (80.0%) were female. Median follow-up time was 52.2 ± 5.8 months (95% confidence interval: 40.7 - 63.7 months). Patients’ demographics and primary disease-related parameters are presented in Table 1. Pre-HSCT MRI/MRA was done in 21 patients. More than half of them had suffered ischemic stroke with moyamoya changes as depicted by MRI evaluation (n = 14, 56.0%), 13 (52.0%) had convulsions and 11 (44.0%) with hemiparesis. Records on pre-HSCT transcranial Doppler (TCD) studies were not available for all the referred patients. Nineteen (76%) patients were receiving monthly packed red blood cell (PRBC) transfusions and iron chelation prior to referral to our institution. Iron overload assessments were done including MRI T2* for those with high serum ferritin.
 Click to view | Table 1. Patient Characteristics and Primary Disease-Related Parameters |
Donors were HLA identical sibling in 21 (84.0%) or identical parent in four (16.0%) recipients. Median cluster of differentiation (CD)34+ cell dose was 5.0 × 106/kg (range: 1.1 - 11.8), whereas total nucleated cell (TNC) dose was 2.98 × 108/kg (range: 1.3 - 45.4). All patients received one to two transfusions during the myelosuppression phase of early transplantation.
Hematopoietic recovery/engraftment/chimerism
ANC recovery was observed in all cases with a median time to ANC recovery of 15 days (range: 12 - 24 days), and platelets recovery was observed in 24 patients with a median recovery time of 26.5 days (range: 16 - 100 days). Sixteen patients (64.0%) exhibited full donor chimerism (100%) in myeloid lineage on day +100 evaluation, while in the remaining recipients it varied from 68% to 96%. There was no event of primary graft failure whereas incidence of secondary graft failure was 4.0% (n = 1) in our cohort. The only patient with secondary graft failure had exhibited 96% chimerism in myeloid line on day +100. Transplanted from an HLA identical brother, the chimerism in myeloid line gradually dropped to 0% for this patient leading to a documented graft failure 14.3 months after infusion. She was on regular transfusions at her last clinical visit for her CV. At the last update to the data set, all but one surviving patients were maintaining sustained donor cells.
GVHD
Cumulative incidence of all grade overall acute GVHD was 20.0% (n = 5), whereas grade I - II acute GVHD accounted for 16% (n = 4) and severe acute GVHD (grade III, gut) was seen in only one case (4.0%). Severe chronic GVHD was seen in three (12.5%) evaluable patients, two of them succumbed to it.
Infections and transplant-related toxicity
In terms of transplant-related morbidity during the day +100, five (25%) recipients had CMV reactivation which was managed successfully using ganciclovir/foscarnet. No other viral infections including Epstein-Barr virus (EBV), adenovirus or BK virus were encountered on routine screening. One patient had hemorrhagic cystitis, and two (8.0%) had sinusoidal obstructive syndrome (SOS); all were self-limiting. None of our patients had transplant-associated thrombotic microangiopathy.
Survival, mortality and causes of death
With a mortality rate of 12.0% (n = 3), all but one of 22 (88%) survivors were hematologically and clinically asymptomatic at the last contact. The cumulative probability of 3-year OS was 92.0% (± 5.4%) and EFS 88.0% (± 6.5%). Causes of death were acute respiratory failure and septic shock due to polymicrobial pneumonia in the first case, acute exacerbation of chronic GVHD of gut in the second patient, and in the third one it was progressive worsening of chronic GVHD of lung (bronchiolitis obliterans); events of death were recorded at 1.6, 6.5, and 45.4 month post-HSCT, respectively.
Post-transplant neurologic assessment
Post-transplant parameters related to neurologic status of the cohort are described in Table 2. Clinically, patients had neurologic examination before and after HSCT regularly in the outpatient clinics and continued on anti-epileptic for 1 year, then it was tapered and discontinued if there were no evidence of epileptic activity on electroencephalogram (EEG). Eleven patients with hemiparesis showed remarkable recovery and regained motor strength (90.9%) except one who continued to have persistent deficit (P = 0.002). Recurrent seizures were seen in 13 patients prior to HSCT; five (38.5%) patients resolved while eight remained to have seizure activity requiring prolonged anticonvulsant therapy. Improvement in neuropsychological abnormality was recorded in three patients and headache resolved in five out of six patients. Post-HSCT radiological imaging was done in 15 patients, which showed stabilization of CV among all. Two of eight patients who had seizures post-HSCT were due to posterior reversible encephalopathy syndrome (PRES) in early post-transplant days. All patients were clinically stable at their last follow-up appointment maintaining normal hematopoiesis, and were leading normal life with no debilitating neurologic deficit except in one who had permanent hemiplegia.
Click to view | Table 2. Post-Transplant Primary Disease Evaluation |
| Discussion | ▴Top |
Our results confirm that HLA-related HSCT offers a curative option and a risk-benefit balance in children who have severe SCD [18-20]. This study is one of a few studies that explored the outcome of HSCT in pediatric SCD patients with CV complications specifically [20]. The incidence of stroke in SCD patients is increased in pediatric age group, which indicates the need for early identification by screening using TCD to detect patients at risk and initiate blood transfusions or refer them for possible HSCT [6, 18, 20]. Unfortunately, most of the patients referred to our center were already symptomatic, due to established vasculopathy and were on regular transfusions. Established CV requires lifelong PRBC transfusions in an attempt to offset the disease progression. However, recurrence of stroke may occur even in patients on transfusion program [17, 22, 23]. For severe symptomatic vasculopathy (moyamoya disease) neurosurgical neo-vascularization EDAS, EMAS can be performed as therapeutic option; however, postoperative complications and recurrence of stroke are usually high [30]. Fourteen of our patients had moyamoya changes on MRI prior to transplantation; all of them stabilized post-HSCT.
Post-HSCT risk of neurological complications has been reported to increase with incidence up to 38% in untransfused patients with prior stroke. Seizures are one of the most common neurological complications [31]. Nevertheless, we found a reduction in seizures in our cohort. Two patients had seizures due to PRES in the early post-transplantation period confirmed with CT scan of the brain. Higher incidence of PRES in SCD (22%) was reported in the literature in patients with SCD following HSCT [29].
HSCT in high-risk SCD patients before development of stroke and CV reduces the risk of subsequent neurologic complications [28]. This was shown using TCD with improved vasculopathy demonstrating low values at 1-, and 3-year follow-up [20]. Extended duration of anticonvulsant post-HSCT with intensified antihypertensive management and platelet support may diminish the frequency of seizures [1, 31]. We also found clinical improvement in our cohort of patients similar to these studies. Considering the high-risk category of our cohort, 3-year OS was 92% and EFS was 88%, still comparable to international reports [18-21]. We also looked at the post-HSCT quality of life for our patients with respect to neurological and psychological status. Our patients showed improvement in their motor and cognitive functions, which was also supported by stabilization of radiological imaging (Table 2, Fig. 1). This report from a single transplant center, will add to the existing literature as it addresses and evaluates specifically the conditions and the outcome of the patients with CV pre- and post-HSCT. Further follow-up of this cohort of patients will continue to be assessed for the long-term effect of HSCT on the neurologic status of these patients. We also have demonstrated in this report a favorable outcome of HSCT in symptomatic patients with severe vasculopathy (moyamoya) compared to those undergoing by-pass surgery EDAS/EMAS for severely narrowed vessels [30].
Limitations
Being a retrospective chart review, our study suffers from obvious limitations. One of the patients was referred to us from another country and had to be censored at 35.6 months at his last follow-up with us. Majority, but not all of the cases had pre- and post-HSCT MRI assessment, importantly, the clinical improvement was remarkable and durable. In addition, TCD studies were not available for all the referred patients prior to HSCT.
Conclusions
HSCT is a curative therapy for high-risk pediatric SCD patients having CV with a favorable outcome resulting in stabilization of brain vascularity and clinical improvement of symptoms like headache, hemiparesis, and convulsion. None of the patients had to go for neo-vascularization procedure before HSCT. Ideally, early TCD should be done to detect high-risk patients followed by early HSCT for eligible patients to avoid the development of neurological complications and to improve the outcome.
Acknowledgments
None to declare.
Financial Disclosure
All authors verify that they do not have any financial disclosure to declare.
Conflict of Interest
The authors declare no conflict of interest.
Informed Consent
Data of interest collected from the patients’ medical records were secured as governed by the institutional policies on patient confidentiality and privacy. No informed consents were obtained since this was a retrospective study and all data items collected were already documented in medical charts as part of the patient care and disease management.
Author Contributions
Abdullah Al-Jefri conceived the idea, reviewed the results, did literature search, prepared the manuscript. Khawar Siddiqui designed the study, performed the statistical analysis of the data and wrote the results for the manuscript. Amira Al-Oraibi collected the data and assisted in literature search. Amal Al-Seraihy, Ali Al Ahmari, Ibrahim Ghemlas, Awatif Al Anazi, Hawazen Al Saedi, and Mouhab Ayas reviewed the results and approved the manuscript.
Data Availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
Abbreviations
SCD: sickle cell disease; HSCT: hematopoietic stem cell transplant; CV: cerebral vasculopathy; PRBC: packed red blood cell; ATG: anti-thymocyte globulin; GVHD: graft-versus-host disease; HLA: human leukocyte antigen; OS: overall survival; EFS: event-free survival; TCD: transcranial Doppler
| References | ▴Top |
- Noe A, Cappelli B, Biffi A, Chiesa R, Frugnoli I, Biral E, Finizio V, et al. High incidence of severe cyclosporine neurotoxicity in children affected by haemoglobinopaties undergoing myeloablative haematopoietic stem cell transplantation: early diagnosis and prompt intervention ameliorates neurological outcome. Ital J Pediatr. 2010;36:14.
doi pubmed - Lehmann H, Maranjian G, Mourant AE. Distribution of sickle-cell hemoglobin in Saudi Arabia. Nature. 1963;198:492-493.
doi pubmed - Al-Qurashi MM, El-Mouzan MI, Al-Herbish AS, Al-Salloum AA, Al-Omar AA. The prevalence of sickle cell disease in Saudi children and adolescents. A community-based survey. Saudi Med J. 2008;29(10):1480-1483.
doi pubmed - Alhamdan NA, Almazrou YY, Alswaidi FM, Choudhry AJ. Premarital screening for thalassemia and sickle cell disease in Saudi Arabia. Genet Med. 2007;9(6):372-377.
doi pubmed - Earley CJ, Kittner SJ, Feeser BR, Gardner J, Epstein A, Wozniak MA, Wityk R, et al. Stroke in children and sickle-cell disease: Baltimore-Washington cooperative young stroke study. Neurology. 1998;51(1):169-176.
doi pubmed - Kirkham FJ, Lagunju IA. Epidemiology of stroke in sickle cell disease. J Clin Med. 2021;10(18):4232.
doi pubmed - Jastaniah W. Epidemiology of sickle cell disease in Saudi Arabia. Ann Saudi Med. 2011;31(3):289-293.
doi pubmed - Fasano RM, Meier ER, Hulbert ML. Cerebral vasculopathy in children with sickle cell anemia. Blood Cells Mol Dis. 2015;54(1):17-25.
doi pubmed - Al Hawsawi ZM, Ismail GA. Stroke among sickle cell disease patients in Madina Maternity and Children's Hospital. Ann Saudi Med. 1998;18(5):472-474.
doi pubmed - Alzahrani F, Fallatah AM, Al-Haddad FM, Khayyat ST, AlMehmadi WM, AlQahtani BG, Alamri RS. Risk factors and complications among pediatric patients with sickle cell anemia: a single tertiary center retrospective study. Cureus. 2021;13(1):e12440.
doi - Brousse V, Kossorotoff M, de Montalembert M. How I manage cerebral vasculopathy in children with sickle cell disease. Br J Haematol. 2015;170(5):615-625.
doi pubmed - Kwiatkowski JL. Matched Sibling Donor Hematopoietic Stem Cell Transplantation to Prevent Stroke in Children With Sickle Cell Anemia. JAMA. 2019;321(3):251-252.
doi pubmed - Abboud MR, Cure J, Granger S, Gallagher D, Hsu L, Wang W, Woods G, et al. Magnetic resonance angiography in children with sickle cell disease and abnormal transcranial Doppler ultrasonography findings enrolled in the STOP study. Blood. 2004;103(7):2822-2826.
doi pubmed - Kandeel AY, Zimmerman RA, Ohene-Frempong K. Comparison of magnetic resonance angiography and conventional angiography in sickle cell disease: clinical significance and reliability. Neuroradiology. 1996;38(5):409-416.
doi pubmed - Powars D, Wilson B, Imbus C, Pegelow C, Allen J. The natural history of stroke in sickle cell disease. Am J Med. 1978;65(3):461-471.
doi - Prengler M, Pavlakis SG, Prohovnik I, Adams RJ. Sickle cell disease: the neurological complications. Ann Neurol. 2002;51(5):543-552.
doi pubmed - Bernaudin F, Socie G, Kuentz M, Chevret S, Duval M, Bertrand Y, Vannier JP, et al. Long-term results of related myeloablative stem-cell transplantation to cure sickle cell disease. Blood. 2007;110(7):2749-2756.
doi pubmed - Gluckman E, Cappelli B, Bernaudin F, Labopin M, Volt F, Carreras J, Pinto Simoes B, et al. Sickle cell disease: an international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood. 2017;129(11):1548-1556.
doi pubmed - Locatelli F, Kabbara N, Ruggeri A, Ghavamzadeh A, Roberts I, Li CK, Bernaudin F, et al. Outcome of patients with hemoglobinopathies given either cord blood or bone marrow transplantation from an HLA-identical sibling. Blood. 2013;122(6):1072-1078.
doi pubmed - Bernaudin F, Verlhac S, Peffault de Latour R, Dalle JH, Brousse V, Petras E, Thuret I, et al. Association of matched sibling donor hematopoietic stem cell transplantation with transcranial doppler velocities in children with sickle cell anemia. JAMA. 2019;321(3):266-276.
doi pubmed - de la Fuente J, Dhedin N, Koyama T, Bernaudin F, Kuentz M, Karnik L, Socie G, et al. Haploidentical bone marrow transplantation with post-transplantation cyclophosphamide plus thiotepa improves donor engraftment in patients with sickle cell anemia: results of an international learning collaborative. Biol Blood Marrow Transplant. 2019;25(6):1197-1209.
doi pubmed - Walters MC, Storb R, Patience M, Leisenring W, Taylor T, Sanders JE, Buchanan GE, et al. Impact of bone marrow transplantation for symptomatic sickle cell disease: an interim report. Multicenter investigation of bone marrow transplantation for sickle cell disease. Blood. 2000;95(6):1918-1924.
- Kamani NR, Walters MC, Carter S, Aquino V, Brochstein JA, Chaudhury S, Eapen M, et al. Unrelated donor cord blood transplantation for children with severe sickle cell disease: results of one cohort from the phase II study from the Blood and Marrow Transplant Clinical Trials Network (BMT CTN). Biol Blood Marrow Transplant. 2012;18(8):1265-1272.
doi pubmed - Panepinto JA, Walters MC, Carreras J, Marsh J, Bredeson CN, Gale RP, Hale GA, et al. Matched-related donor transplantation for sickle cell disease: report from the Center for International Blood and Transplant Research. Br J Haematol. 2007;137(5):479-485.
doi pubmed - Dallas MH, Triplett B, Shook DR, Hartford C, Srinivasan A, Laver J, Ware R, et al. Long-term outcome and evaluation of organ function in pediatric patients undergoing haploidentical and matched related hematopoietic cell transplantation for sickle cell disease. Biol Blood Marrow Transplant. 2013;19(5):820-830.
doi pubmed - Walters MC, Patience M, Leisenring W, Eckman JR, Scott JP, Mentzer WC, Davies SC, et al. Bone marrow transplantation for sickle cell disease. N Engl J Med. 1996;335(6):369-376.
doi pubmed - Hsieh MM, Fitzhugh CD, Weitzel RP, Link ME, Coles WA, Zhao X, Rodgers GP, et al. Nonmyeloablative HLA-matched sibling allogeneic hematopoietic stem cell transplantation for severe sickle cell phenotype. JAMA. 2014;312(1):48-56.
doi pubmed - Walters MC, Hardy K, Edwards S, Adamkiewicz T, Barkovich J, Bernaudin F, Buchanan GR, et al. Pulmonary, gonadal, and central nervous system status after bone marrow transplantation for sickle cell disease. Biol Blood Marrow Transplant. 2010;16(2):263-272.
doi pubmed - Gaziev J, Marziali S, Paciaroni K, Isgro A, Di Giuliano F, Rossi G, Marziali M, et al. Posterior reversible encephalopathy syndrome after hematopoietic cell transplantation in children with hemoglobinopathies. Biol Blood Marrow Transplant. 2017;23(9):1531-1540.
doi pubmed - Griessenauer CJ, Lebensburger JD, Chua MH, Fisher WS, 3rd, Hilliard L, Bemrich-Stolz CJ, Howard TH, et al. Encephaloduroarteriosynangiosis and encephalomyoarteriosynangiosis for treatment of moyamoya syndrome in pediatric patients with sickle cell disease. J Neurosurg Pediatr. 2015;16(1):64-73.
doi pubmed - Walters MC, Sullivan KM, Bernaudin F, Souillet G, Vannier JP, Johnson FL, Lenarsky C, et al. Neurologic complications after allogeneic marrow transplantation for sickle cell anemia. Blood. 1995;85(4):879-884.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.