

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website http://www.thejh.org |
Original Article
Volume 8, Number 1, March 2019, pages 11-16
Trends in Hospitalizations for Sickle Cell Disease Related-Complications in USA 2004 - 2012
An Thi Nhat Hoa, c, Artem Shmelevb, Astha Joshia, Nghi Hoa
aMedStar Harbor Hospital, Baltimore, MD, USA
bSaint Agnes Hospital, Baltimore, MD, USA
cCorresponding Author: An Thi Nhat Ho, MedStar Harbor Hospital, Baltimore, MD 21225, USA
Manuscript submitted January 4, 2019, accepted February 26, 2019
Short title: Hospitalizations for SCD Related-Complications
doi: https://doi.org/10.14740/jh475
| Abstract | ▴Top |
Background: Sickle cell disease (SCD) affects 100,000 patients in the USA. However, no recent data was available for annual national trends in hospitalization rates, in-hospital mortality, hospital length of stay (LOS) and costs of SCD admissions due to its complications.
Methods: This study was conducted to study the trends of hospitalization rates, in-hospital mortality, LOS and hospital charges due to SCD-related complications in African American (AA) patients from 2004 to 2012 in the USA. Complications included acute chest syndrome, splenic sequestration, bacterial pneumonia, sepsis, stroke, deep vein thrombosis (DVT) or pulmonary embolism, retinal circulation complications, priapism, disorders related to biliary stones, or those required blood transfusions. We obtained the study population from the Nationwide Inpatient Sample.
Results: Hospital admission rate rose steadily from 106 per 100,000 AA population in 2004 to 137 in 2012. Seasonal and trend decomposition revealed the highest hospitalization rate in January. Hospital LOS decreased from 7.1 ± 7.65 days in 2004 to 6.23 ± 6.42 days in 2012. Hospital charges increased from 15.35 (8.99 - 27.57) thousand dollars per admission in 2004 to 24.78 (14.37 - 45.24) in 2012. Medicaid remained the primary payer in the highest number of patients in 9 years. In-hospital mortality did not change significantly, being 1.03% in 2004 and 1.02% in 2012, with no significant seasonal variation in mortality. Most common complications were acute pain crisis and blood transfusion requirement. Biliary pathology was the only complication that decreased over time. Admissions for each complication were initially uprising with a decline from 2010 to 2012, except for DVT/pulmonary embolism with a significant uptrend.
Conclusions: Overall, from 2004 to 2012, hospital admission rates and charges increased, and hospital LOS decreased, while in-hospital mortality remained unchanged.
Keywords: Sickle cell disease; Trend; In-hospital mortality; Admission rates; Length of stay
| Introduction | ▴Top |
Sickle cell disease (SCD) is a common genetic disorder that causes hemolysis and superimposed acute and chronic complications by vaso-occlusion, vascular injury, and ultimately organ damage leading to death. It affects more than 100,000 individuals in the USA, the majority of whom are African-American (AA) [1]. With the recent advancements in the treatment and screening, there is some evidence that survival of patients with SCD has improved [2]. In 1973, the estimated mean age at death of SCD patients in the USA was 14.3 years, with 50% of deaths occurring during the first 5 years of life [3]. By early 1990s, the cooperative study of SCD estimated a median life expectancy of those with sickle cell anemia, the most severe form of the disease, of 42 years for men and 48 years for women [4]. Incidence of complications, such as stroke in children with SCD, has also decreased [5]. This dramatic improvement has been attributed to several interventions starting in early childhood, including widespread newborn screening programs, the use of penicillin prophylaxis, hydroxyurea, and pneumococcal vaccination. However, population-based analysis from 1998 to 2008 did not show a steady decrease in hospitalization rates of sickle cell patients [6] and studies of most recent hospitalization trends are not yet available. The objective of our study was to describe annual national trends in hospitalization rates, in-hospital mortality, hospital length of stay (LOS) and costs of SCD admissions due to its complications from 2004 to 2012, using large administrative data set.
| Methods | ▴Top |
We obtained the study population from Nationwide Inpatient Sample (NIS) of Agency for Healthcare Research and Quality (AHRQ) Healthcare Cost and Utilization Project (HCUP), from 2004 to 2012. Appropriate weighting was used to produce accurate nation-wide estimates.
Study population was limited to AA of any age, admitted with any of International Classification of Diseases, Ninth Revision, Clinical Modification (ICD-9-CM) diagnostic codes for SCD, with or without evidence of crisis, who additionally had documented ICD-9-CM codes for SCD-related complications (acute chest syndrome, splenic sequestration, bacterial pneumonia, cerebro-vascular accidents, deep vein thrombosis (DVT) or pulmonary embolism, retinal circulation complications, priapism, disorders related to biliary stones, sepsis, or those required blood transfusions). Annual estimates of AA population were provided by U.S. Census Bureau, to account for growing US population and to produce hospitalization rates. Corresponding ICD-9-CM codes can be found in Supplementary 1 (http://www.thejh.org/index.php/jh).
Next, weighted annual and monthly admission rates, annual and monthly mortality rates, values of LOS and hospital charges were calculated. Monthly hospitalization rates or metrics represent a time series and can be therefore analyzed with special tools, as seasonal and trend decomposition.
Time series seasonal and trend decomposition was performed to split original rates into trend component (representing long-term yearly changes), seasonal component (circannual changes), and residual, or random, component (noise). Locally estimated scatterplot smoothing (LOESS) was selected as a decomposition method as it provided minimal autocorrelation of residual component of the time series, which proves absence of hidden, undetected patterns.
| Results | ▴Top |
Admission rates and characteristics
Annual characteristics of hospitalizations for SCD complications are summarized in Table 1. We observed a 30% rise in hospital admissions from 39,802 admissions (106 per 100,000 AA population) in 2004 to 56,635 admissions (137 per 100,000 AA population) in 2012 (Fig. 1). There was a steady uptrend from 106 per 100,000 AA population to 157 in 2010, but then it decreased slightly from 157 in 2010 to 137 in 2012. Female proportion was higher than male proportion in all the years (58.7% vs. 41.3%) with an average age of 33.38 ± 11.70. Over years, the average age of hospitalized sickle cell patients did not demonstrate clinically significant difference; however, it reached statistical significance due to large sample size.
 Click to view | Table 1. Annual Hospitalization Rates, Demographics, Hospital-Level Characteristics, Mortality, LOS and Charges |
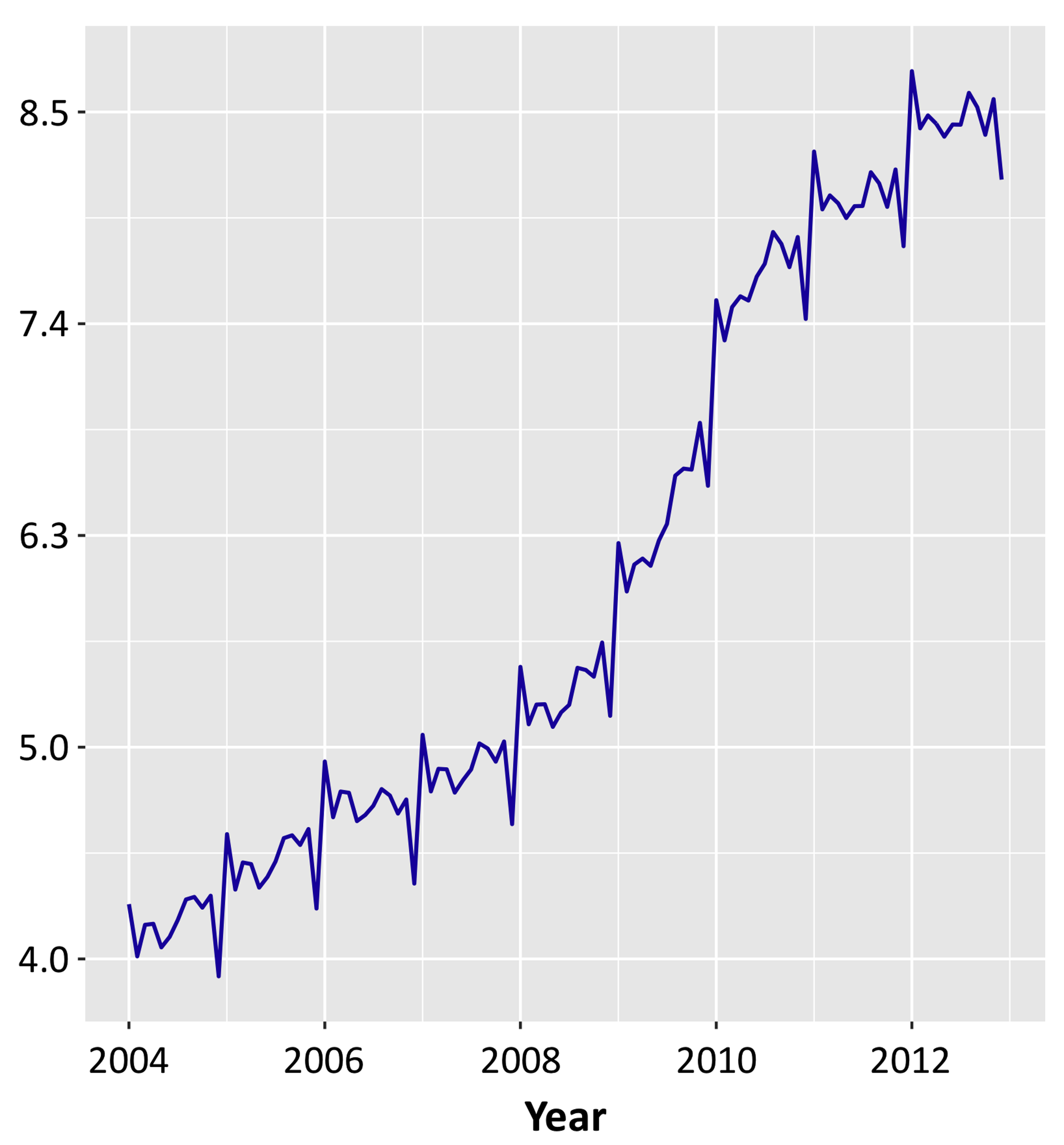 Click for large image | Figure 1. Monthly incidence of hospitalizations for SCD complications, per 100,000 African-American population. SCD: sickle cell disease. |
Number of admissions increased the most in the West region (2-fold increase from 2004 to 2012 compared to 1.3 - 1.4 in North East, South and West).
Seasonal decomposition revealed relatively uniform admission rate throughout the year with exception for January, with the highest hospitalization rate, and December, with significant trough (Fig. 2).
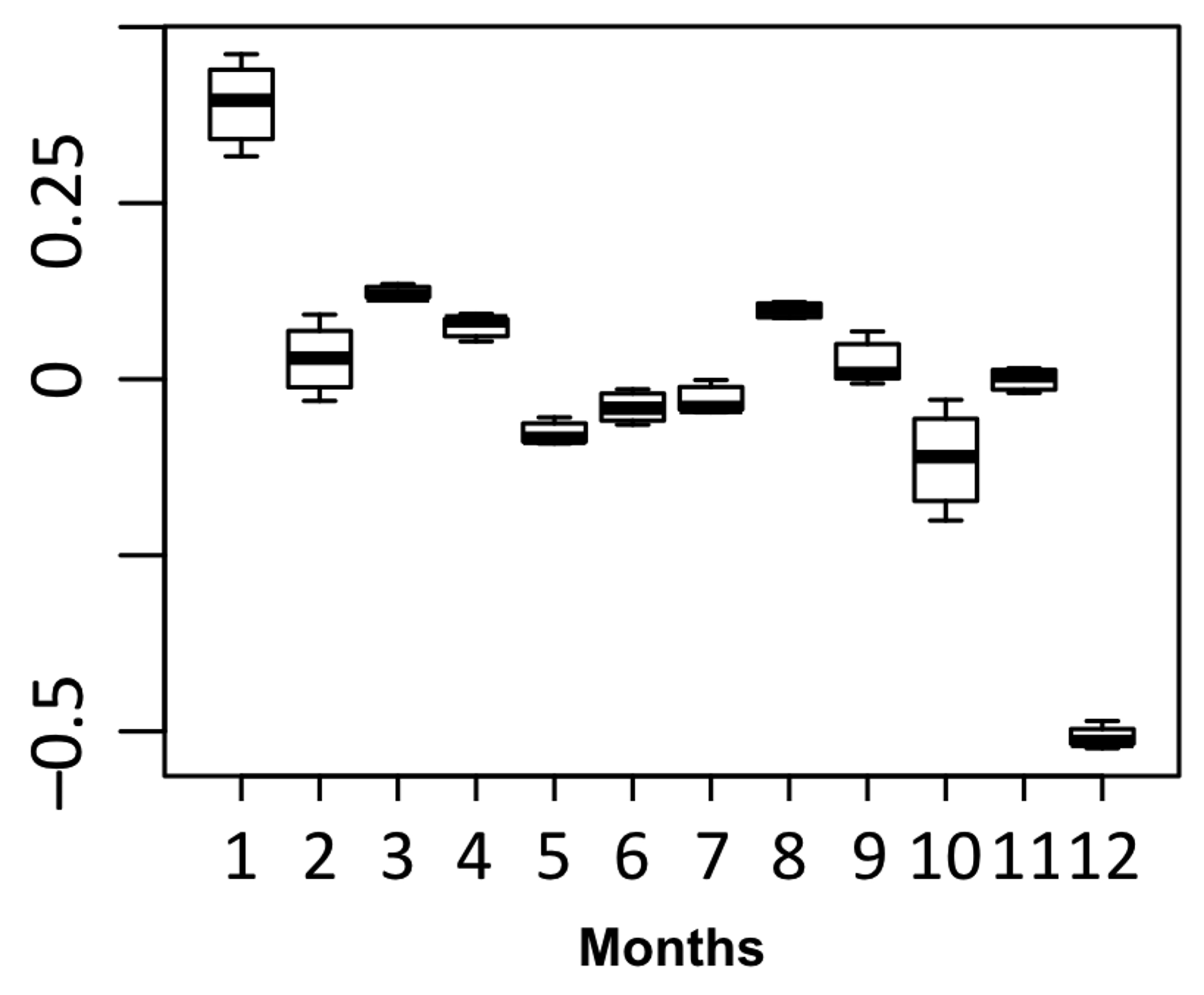 Click for large image | Figure 2. Seasonal deviation in monthly hospitalization rates, per 100,000 AA population. AA: African-American. |
Complication rate
Pure complications (i.e. present given complication with absence of other complications) were analyzed (Fig. 3). Acute pain crisis and anemia requiring blood transfusion were the most common reasons for admission in 2004 - 2012 time span (374,335 and 222,411 admissions respectively), followed by DVT, sepsis, and biliary tract disorders. Overall, the trend of admission rate per 100,000 AA population for each complication increased from 2004 to 2010 except for biliary pathology. From 2010 to 2012, admission rate for all complications started to decline, except for priapism, DVT and disturbances of retinal circulation. The results are almost similar when we counted admissions for individual complications but with other overlapping complications.
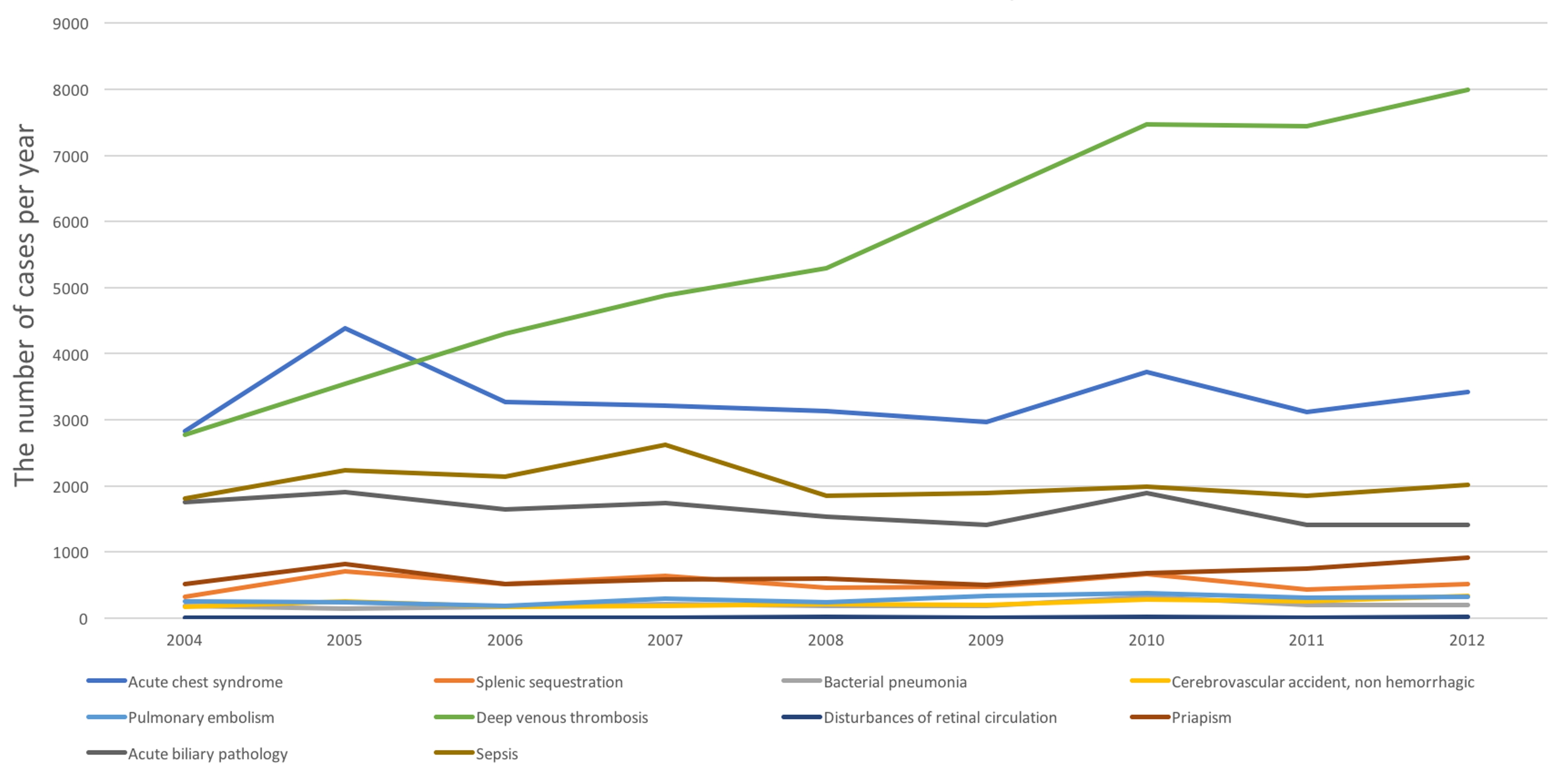 Click for large image | Figure 3. Trend in number of admissions for individual complications. |
Average LOS, costs, and in-hospital mortality
Over the period of 9 years, average LOS of admissions for any complications decreased from 7.12 ± 7.65 days in 2004 to 6.23 ± 6.42 days in 2012 (P < 0.001).
The average total charges of each hospital stay increased by 1.5 times from 25.36 (95% confidence interval (CI) 8.99 -27.57) in 2004 to 41.32 (95% CI 14.37 - 45.24) in 2012. This increase was monotonous with no declines.
Absolute mortality rates did not change in clinically significant way, being 1.03% in 2009, and with minimal value of 0.84% in 2010, and 1.02% in 2012 (Fig. 4).
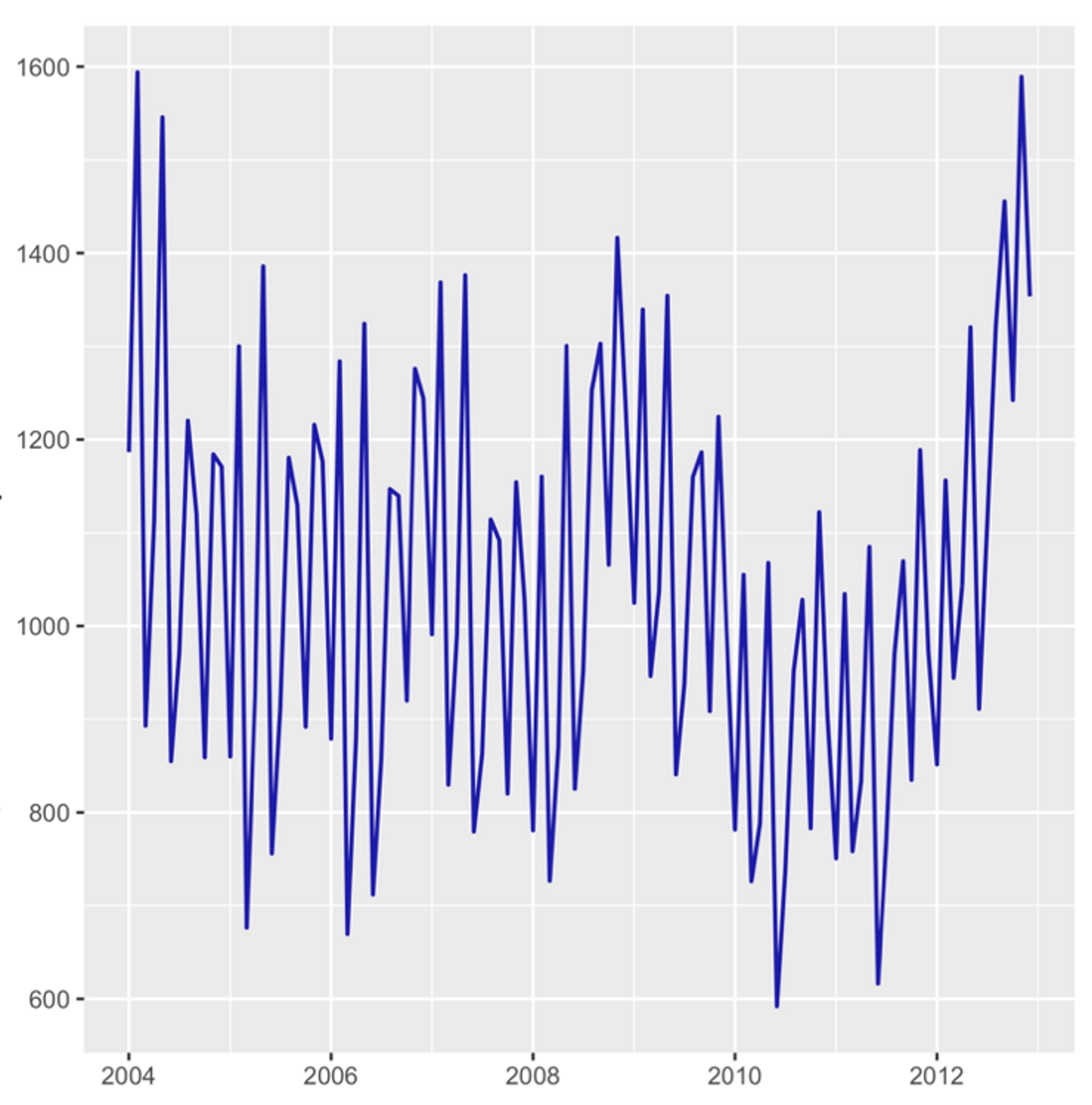 Click for large image | Figure 4. Seasonal deviation in in-hospital mortality rate, per 100,000 hospitalizations. |
| Discussion | ▴Top |
To the best of our knowledge, our study is the first to examine the most recent longitudinal data for hospitalizations due to complications of SCD. Our study found an increase in admission rate in adult AA population up to about 30% in the period from 2004 to 2012. In an era of advances in complication prevention and treatment of SCD, especially since hydroxyurea was approved by Food and Drug Administration (FDA) in 1996, this increase in complication-related admissions could be explained by improvement in SCD diagnostic rate by implementation of state-wide newborn screening programs in the early 1980s - 1990s [7]. Another possibility is that the patients with advances in treatment now have a longer life expectancy and are admitted more frequently for SCD complications. Okam et al in their NIS study [6] also found a statistically non-significant fluctuation in general admissions with a diagnosis of SCD in a period from 1998 to 2008.
Interestingly, based on seasonal decomposition, observed hospitalization rate was highest in January with significant decline in December. The rest of the years were characterized by relatively uniform admission rate with mild inter-month fluctuations. This pattern occurred independently of the year. Smith et al observed this pattern in a study of 299 patients and found that sickle cell pain intensity usually peaked in late fall/early winter and troughed in spring. Patients with SCD exhibit hypersensitivity to thermal stimuli [8] and often report cooler weather or exposure to cold as the most important precipitating factor for painful vaso-occlusive crises (VOCs) [9-11]. The reflex constriction of superficial blood vessels in response to skin cooling is enhanced in SCD as compared to normal individuals. Serjeant and Chalmers hypothesized that this vasoconstriction may be associated with diversion of blood (“vascular steal”) away from active bone marrow and may cause avascular necrosis and precipitate VOC [12].
Of all the causes of admissions, acute pain crisis and anemia requiring blood transfusion topped among all other complications. Biliary pathology related admissions were the only one that decreased over time. Admission rates for all other complications increased but started to stabilize from 2010 to 2012, except DVT which significantly increased over 8 years. Although hydroxyurea seems to decrease coagulation markers, its effect on venous thrombosis is still unclear [13, 14]. In general population, despite the advances in pharmacotherapy, admission rates for DVT did not decrease in a 10-year period [15]. In SCD patients who harbor SCD-related risk factors as well as frequent hospitalizations for pain and complications, our finding of increased admission rate for DVT could be explained.
Overall, there was a decrease in average LOS. However, in-hospital mortality associated with patients admitted for complications of SCD did not change significantly over 9 years from 2004 to 2012. Since hydroxyurea was first approved for adult patients with SCD in 1998, its long-term use has been proved to prevent and shorten hospital duration of stay due to complications and improve survival [3, 16] but data on whether this medication has effect on in-hospital mortality is limited. In addition, advances in therapy for SCD including immunomodulatory drugs, DNA-methyltransferase inhibitors and hematologic stem cell transplant were not widely implemented in the period of 2004 - 2012. The fact of decreased LOS with increased admission rate also raised questions about possible readmission rate which was beyond the scope of our study.
Our results showed a tremendous increase in hospital cost over the time frame of 9 years. Increasing disposition, greater complexity of treatments, and ageing population are some causes of this hospital cost increase. The financial side of healthcare delivery has been more and more of a staggering burden for the economy.
Limitations
Although we have included all reasonable ICD-9-CM codes for SCD and its complications, our study was based on nosology units which has billable codes, and thus is susceptible to errors arising from coding inaccuracies. Second, we attempted to include all the complications that are possibly contributed by SCD but in fact, this could be related to other comorbidities of individual patients as well. This could result in an overestimation of admissions due to complications only related to SCD. The database we used also allowed us to study only in-hospital mortality but did not have value in follow-up outcomes. Nevertheless, this study has provided with an important and objective general overview of a large sample of hospitalized SCD patients over a relatively long period of time.
Conclusions
From 2004 to 2012, admission rate for SCD-related complications increased. Most common complications were acute pain crisis and blood transfusion requirement. Amongst all the complications, biliary pathology related admissions decreased while all other complications increased with DVT rising remarkably. Average length of hospital stay decreased during this period with an increased trend in hospital charges. However, in-hospital mortality remained unchanged.
Conflict of Interest
The authors have no conflict of interest to disclose.
| References | ▴Top |
- Hassell KL. Population estimates of sickle cell disease in the U.S. Am J Prev Med. 2010;38(4 Suppl):S512-521.
doi pubmed - Paulukonis ST, Eckman JR, Snyder AB, Hagar W, Feuchtbaum LB, Zhou M, Grant AM, et al. Defining sickle cell disease mortality using a population-based surveillance system, 2004 through 2008. Public Health Rep. 2016;131(2):367-375.
doi pubmed - Steinberg MH, McCarthy WF, Castro O, Ballas SK, Armstrong FD, Smith W, Ataga K, et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: A 17.5 year follow-up. Am J Hematol. 2010;85(6):403-408.
doi - Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, Klug PP. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330(23):1639-1644.
doi pubmed - McCavit TL, Xuan L, Zhang S, Flores G, Quinn CT. National trends in incidence rates of hospitalization for stroke in children with sickle cell disease. Pediatr Blood Cancer. 2013;60(5):823-827.
doi pubmed - Okam MM, Shaykevich S, Ebert BL, Zaslavsky AM, Ayanian JZ. National trends in hospitalizations for sickle cell disease in the United States following the FDA approval of hydroxyurea, 1998-2008. Med Care. 2014;52(7):612-618.
doi pubmed - Yusuf HR, Lloyd-Puryear MA, Grant AM, Parker CS, Creary MS, Atrash HK. Sickle cell disease: the need for a public health agenda. Am J Prev Med. 2011;41(6 Suppl 4):S376-383.
doi pubmed - Brandow AM, Stucky CL, Hillery CA, Hoffmann RG, Panepinto JA. Patients with sickle cell disease have increased sensitivity to cold and heat. Am J Hematol. 2013;88(1):37-43.
doi pubmed - Baum KF, Dunn DT, Maude GH, Serjeant GR. The painful crisis of homozygous sickle cell disease. A study of the risk factors. Arch Intern Med. 1987;147(7):1231-1234.
doi pubmed - Murray N, May A. Painful crises in sickle cell disease - patients' perspectives. BMJ. 1988;297(6646):452-454.
doi pubmed - Serjeant GR, Ceulaer CD, Lethbridge R, Morris J, Singhal A, Thomas PW. The painful crisis of homozygous sickle cell disease: clinical features. Br J Haematol. 1994;87(3):586-591.
doi pubmed - Serjeant GR, Chalmers RM. Current concerns in haematology. 1. Is the painful crisis of sickle cell disease a "steal" syndrome? J Clin Pathol. 1990;43(10):789-791.
doi - Colella MP, De Paula EV, Conran N, Machado-Neto JA, Annicchino-Bizzacchi JM, Costa FF, Saad ST, et al. Hydroxyurea is associated with reductions in hypercoagulability markers in sickle cell anemia. J Thromb Haemost. 2012;10(9):1967-1970.
doi pubmed - Naik RP, Streiff MB, Lanzkron S. Sickle cell disease and venous thromboembolism: what the anticoagulation expert needs to know. J Thromb Thrombolysis. 2013;35(3):352-358.
doi pubmed - Mansour S, Alotaibi G, Wu C, McMurtry MS. Trends in admission rates and in-hospital stay for venous thromboembolism. Thromb Res. 2017;156:149-154.
doi pubmed - Steinberg MH, Barton F, Castro O, Pegelow CH, Ballas SK, Kutlar A, Orringer E, et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. JAMA. 2003;289(13):1645-1651.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.